不同驅動力條件下改性淀粉、羧甲基纖維素鈉和黃原膠對水合物形成的影響
2021-03-03 07:27:44孫金聲孫慧翠史曉梅王金堂郭東東
中國石油大學學報(自然科學版) 2021年1期
王 韌, 孫金聲,, 孫慧翠, 張 凌,史曉梅, 王金堂, 郭東東, 張 潔
(1.中國石油集團工程技術研究院有限公司,北京 102206; 2.非常規油氣開發教育部重點實驗室(中國石油大學(華東)),山東青島 266580; 3.中國石油大學(華東)石油工程學院,山東青島 266580; 4.中國地質大學(武漢)工程學院,湖北武漢 430074)
天然氣水合物是一種潛力巨大的新型替代能源[1]。凍土及海洋區域天然氣水合物的勘探與開發備受關注[2-3]。水合物鉆井過程中淺層氣及鉆穿儲層時水合物的分解氣會侵入鉆井液[5-6],在改變鉆井液性能的同時還促使水合物形成和聚集于井內,進而誘發安全隱患[5-8]。水合物鉆井為抑制儲層中水合物的分解,需對井內泵入溫度較低的鉆井液[9],但低溫鉆井液進入井筒后會與井下鉆具及地層進行熱量交換,致使其始終處于變溫狀態,而鉆井液溫度的變化又會對其流變性產生影響[9],所以水合物鉆井液在具有良好水合物抑制性的同時,其流變性在較大幅度的變溫條件下需兼具良好的可控性,而這又與所選用的增黏劑密切相關[10]。對于鉆井液增黏劑對水合物形成影響[11-15],多數研究是在某一個驅動力條件下進行鉆井液增黏劑的水合物抑制性評價,筆者在不同實驗條件下研究加量(質量分數)為0.1%~0.5%的改性淀粉、羧甲基纖維素鈉(CMC)和黃原膠(XC)對CH4水合物形成的影響,結合溶液介觀結構特征,采用多種實驗方法、多角度揭示影響機制;綜合考慮3種增黏劑的提黏效果及變溫條件下(5~20 ℃)水溶液流變性的變化,分析3種增黏劑在不同鉆井液體系中的適用情況。
1 實 驗
1.1 實驗材料與裝置
實驗材料包括中軒生化股份有限公司生產的改性淀粉和XC、國藥集團化學試劑有限公司生產的高黏CMC、武漢紐瑞德特種氣體有限公司生產的CH4氣體(純度高于99.9%)以及實驗室自制蒸餾水。水合物形成模擬實驗使用HCSHW-Ⅰ型多功能水合物反應模擬裝置完成(圖1);介觀結構觀測實驗需先利用LGJ-10D型冷凍干燥機制備溶液凍干樣品,再利用Phenom pro型臺式掃描電子顯微鏡進行觀測。
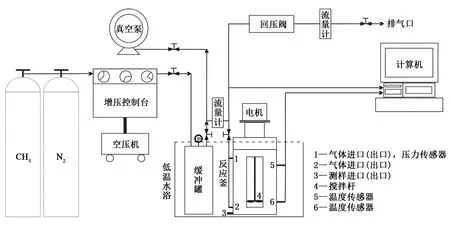
圖1 實驗裝置示意圖Fig.1 Schematic of experimental setup
1.2 實驗方法與過程
1.2.1 CH4水合物形成
水合物鉆井通常使用溫度較低的鉆井液體系[9],而低溫條件下鉆井液的黏度相對較高[16]。選擇質量分數較低的增黏劑水溶液(0.1%~0.5%)作為研究對象,各組溶液均在600 r/min的轉速下攪拌2 h制得。
水合物及常規深海油氣鉆井過程中,循環狀態下的鉆井液所處溫壓環境持續變化,從而使得發生氣侵時水合物形成的驅動力條件也會有所變化。參考純水體系中CH4水合物相平衡條件(5 ℃、4.56 MPa[17])及中國南海水合物勘探項目測得的井內溫壓條件[18],對5 ℃、5 MPa(水合物形成過冷度約為1.3 ℃,驅動力較弱)和5 ℃、12 MPa(水合物形成過冷度約為10.1 ℃,驅動力較強)這2個初始實驗條件下,不同種類及加量的增黏劑對CH4水合物形成的影響進行研究。為確保實驗的可重復性,每組實驗均重復3次。
(1)清洗容積為650 mL的反應釜并檢驗其氣密性;
(2)釜內抽真空的同時打開測樣進出口將250 mL待測液樣吸入釜內,裝樣完畢后持續抽真空30 min;
(3)向緩沖罐內注入CH4氣體直至壓力達到20 MPa;
(4)啟動溫控系統對實驗體系進行控溫,開啟機械攪拌使液樣在300 r/min動態條件下均勻降溫;
(5)實驗體系溫度降至并穩定在5 ℃后,打開反應釜上進氣口使緩沖罐內CH4氣體進入釜內直至壓力達到實驗設計值;
(6)打開監測軟件采集釜內溫壓數據,并將機械攪拌轉速增至600 r/min。
1.2.2 增黏劑水溶液結構
了解增黏劑水溶液的介觀結構特征有助于更好地認識和揭示其對水合物形成影響的內在機制[19]。對各組溶液進行冷凍干燥制樣,再利用掃描電鏡觀測其介觀結構:
(1)使用滴管移取少量溶液滴至新解理的云母片上,然后放入液氮中冷凍2 min;
(2)將冷凍完成的樣品放入凍干機進行12 h的冷凍干燥;
(3)樣品制備完成后利用掃描電鏡進行觀測。
1.3 實驗數據的處理
依據實驗過程中反應釜內溫度(氣、液兩相)和壓力隨時間的變化可得到相應的溫壓曲線,進而從溫壓曲線上讀取水合物開始、停止形成的時間以及這兩個時間點釜內的壓力值等重要參數。
評價鉆井液水合物抑制性時,需從水合物形成的誘導時間、形成量、形成速率等角度進行全面的分析[20]。在此前提下,采用廣義方法對水合物形成誘導時間進行判定[21];由于實驗所用客體氣體為純度較高的CH4氣體,所以其消耗量代表了水合物的形成量,氣體消耗量計算公式[22]為
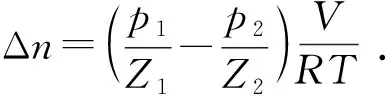
(1)
式中,Δn為氣體消耗量,mol;R為氣體常數,8.314 J/(mol·K);T為氣體溫度,K;V為氣體體積,m3;p1和p2分別為水合物開始和停止形成時釜內壓力,Pa;Z1和Z2分別為p1、p2狀態下氣體的壓縮因子,使用天然氣偏差系數計算軟件計算得出。
水合物形成速率通過水合物開始形成至停止形成這段時間內CH4氣體的平均消耗速率進行表征,計算公式為
(2)
式中,v為氣體消耗速率,mol/min;t1和t2分別為水合物停止形成時間和誘導時間,min。
2 實驗結果
2.1 增黏劑水合物抑制性評價
2.1.1 初始溫壓條件為5 ℃、5 MPa
圖2為水合物形成前—中—后期的溫壓曲線。計算得出各組實驗水合物形成的誘導時間、形成量(氣體消耗物質的量)及形成速率(氣體消耗平均速率)見圖3。
分析圖2及圖3可知,與蒸餾水實驗相比各組增黏劑水溶液均具有不同程度的水合物抑制性。改性淀粉水溶液中水合物形成誘導時間和形成速率均隨溶液質量分數的增大逐漸延長和減慢,但水合物形成量卻相差極小且與蒸餾水實驗基本相同。CMC水溶液質量分數為0.1%和0.2%時,水合物形成誘導時間及形成速率的變化趨勢與改性淀粉類似,但變化幅度都相對更大,水合物的形成量同樣與蒸餾水實驗相近;當溶液質量分數達到及超過0.3%后,標志著水合物形成的溫度驟升和壓力驟降均未出現,且整個實驗過程中CH4氣體消耗量可以忽略不計(約0.003 mol),所以判定這3個質量分數條件下體系內基本無水合物形成。XC水溶液質量分數由0.1%增至0.3%的過程中,水合物形成誘導時間、形成量及形成速率的變化趨勢與CMC實驗相似,但質量分數為0.1%和0.2%時水合物形成的誘導時間相對略長;質量分數增大到0.4%和0.5%后,體系中出現約0.024和0.043 mol的氣體消耗。
綜上所述,水合物形成驅動力較弱時,雖然改性淀粉抑制水合物形成和生長的能力隨加量的增大逐漸增強,但水合物抑制性相對較弱;CMC對水合物形成和生長的抑制能力則相對較強,且加量達到或超過0.3%后能夠徹底抑水合物的形成;XC的水合物抑制性同樣相對較強,加量為0.3%時也能夠徹底抑制水合物的形成,但隨著加量的繼續增大體系中又會形成少量的水合物,因此XC的加量為0.3%時水合物抑制性最強。
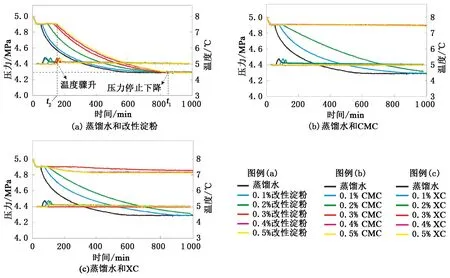
圖2 驅動力較弱時各組實驗溫度和壓力隨時間變化Fig.2 Changes in temperature and pressure in each experiment when driving force is weak
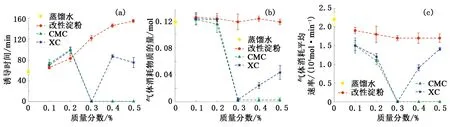
圖3 驅動力較弱時水合物形成誘導時間、形成量、形成速率隨溶液質量分數的變化Fig.3 Hydrate formation induction time, amount and rate change with mass fraction of solution when driving force is weak
2.1.2 初始溫壓條件為5 ℃、12 MPa
圖4中為水合物形成過程中的溫壓曲線。由圖4可知,各組實驗開始后釜內壓力驟降的同時溫度出現驟升的情況,說明水合物形成的誘導時間極短甚至沒有;由于各組實驗的初始壓力和最終壓力幾乎相同,所以水合物的形成量也基本相同,但各組實驗水合物停止形成的時間存在差異,使得水合物的形成速率有所不同(圖5)。
由圖4和圖5可知,3種增黏劑加量均為0.1%時,各組實驗水合物形成速率略慢于蒸餾水實驗,說明3種增黏劑在此加量下水合物抑制性較弱。改性淀粉加量增至0.2%后,其水合物抑制性幾乎沒有變化,但此質量分數條件下CMC和XC水溶液中的溫壓曲線卻與蒸餾水的有些偏離,水合物形成速率繼續減慢,CMC和XC的水合物抑制性有所增強,且XC相對更強。3種增黏劑水溶液質量分數達到0.3%~0.5%后,水合物形成速率皆隨濃度增大出現較明顯的減慢,且增黏劑加量相同時改性淀粉的水合物抑制性最弱,XC略強于CMC。
驅動力較強的情況下,雖然3種增黏劑抑制水合物成核和生長的能力會隨加量的增大逐漸增強,但抑制作用相對較弱,僅在一定程度上減慢了水合物的形成速率。對比可知,此實驗條件下XC的水合物抑制性相對而言是最強的。
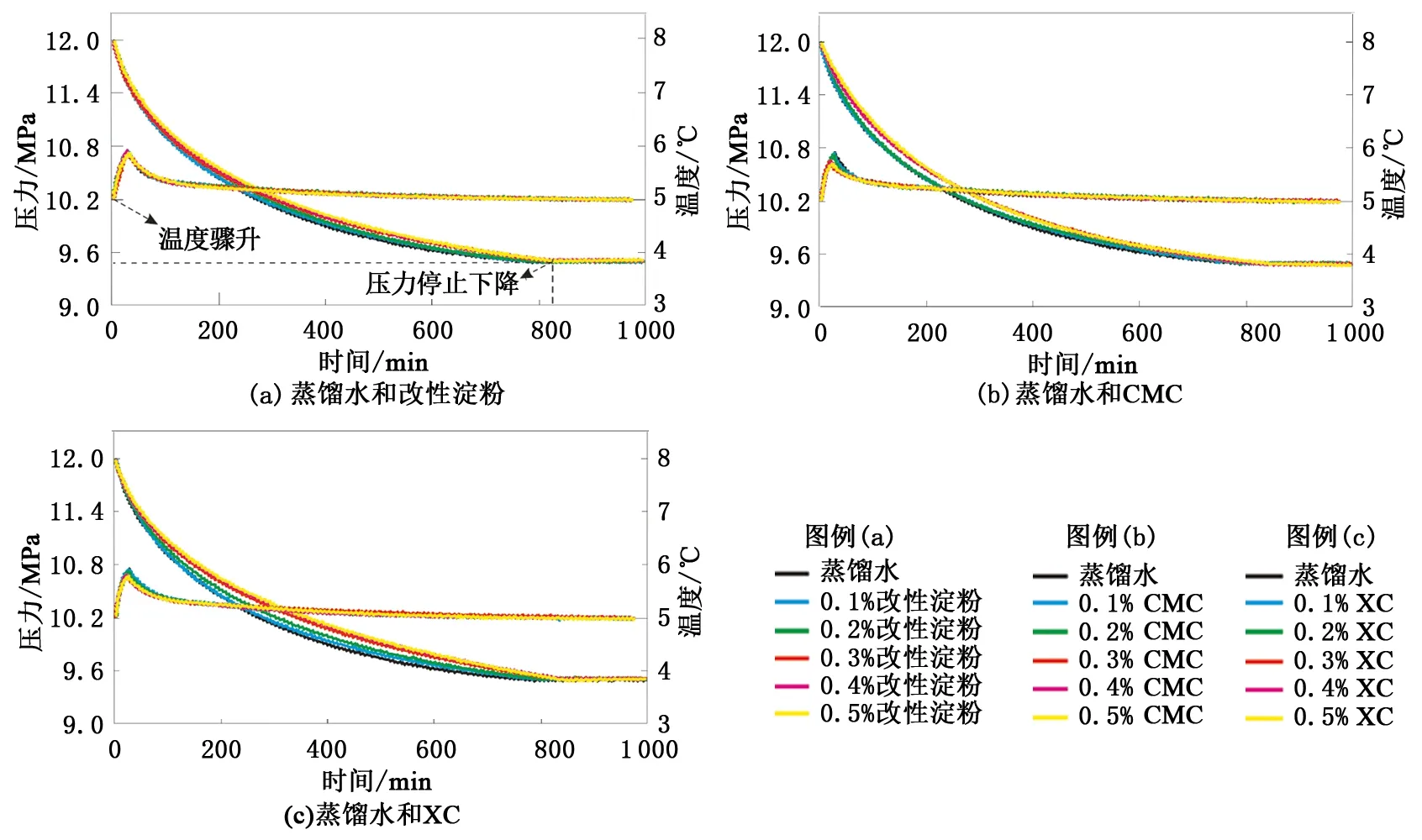
圖4 驅動力較強時各組實驗溫度和壓力隨時間變化Fig.4 Changes in temperature and pressure in each experiment when driving force is strong

圖5 驅動力較強時水合物形成速率隨溶液質量分數的變化Fig.5 Hydrate formation rate changes with mass fraction of solution when driving force is strong
2.2 介觀結構觀測
圖6為各凍干樣品不同放大倍數下的介觀結構,其中亮白色部分為增黏劑水化分子聚集體,暗黑色部分為孔隙空間。觀察圖6可知,增黏劑水溶液質量分數為0.3%時,改性淀粉水化分子組成的空間結構主要由分布較為稀疏的桿狀網絡骨架(直徑約0.5~3 μm)組成,網絡骨架之間較密集地分布著直徑約5~25、50~80和100~200 μm的孔隙,且孔隙通道較大(直徑約50~100 μm)、連通性較好;CMC水化分子聚集體的空間結構主要由分布較為均勻的光滑薄片狀骨架(厚度約0.2 μm,彼此間距約20~50 μm)與稀疏分布于它們之間的線狀骨架(直徑約為0.5 μm)組成,薄片狀骨架之間的孔隙空間相對改性淀粉的要小一些(孔隙通道直徑約為5~50 μm)且形態較復雜;XC水化分子聚集體的空間結構與CMC類似,但薄片狀骨架更薄(厚度約為0.1 μm)、彼此間距更小(約為10~50 μm),支撐薄片狀骨架的線狀骨架更細(直徑約為0.3 μm)、分布更密集,使得網絡空間孔隙更小(孔隙通道直徑約為2~30 μm)且形態更復雜。隨著溶液中3種增黏劑加量的逐漸升高,由增黏劑水化分子構成的網絡骨架變厚、變粗,并且孔隙空間尺寸與形態也隨之變小、變復雜。綜上所述,3種增黏劑在水中所構成網絡骨架的空間結構存在較大差異,而這些差異極有可能影響并決定了它們的水合物抑制性[19]。
3 分析與討論
3.1 增黏劑水合物抑制性及抑制機制
水合物形成驅動力較弱時,實驗開始后幾乎相同量的CH4氣體溶解于各組溶液,由于改性淀粉水溶液中存在的均為分布稀疏的桿狀骨架,所以其水化分子聚集體的比表面積相對較小,并且由于較大孔隙空間的存在,親水的改性淀粉水化分子附近所束縛的水分子相對較少,孔隙空間中的自由水分子相對較多[23-24]。疏水的非極性CH4分子溶解于溶液中后很可能較多的分散于自由水中[25],使得孔隙空間中氣液接觸面積相對較大,容易在氫鍵作用下形成水合物,所以改性淀粉的水合物抑制性相對較弱。隨著加量的增大,雖然改性淀粉水化分子聚集體的比表面積有所增大,骨架結構中的孔隙空間有所減小,自由水分子的數量有所減少,但孔隙空間中很可能仍存在較大量的自由水分子,使得改性淀粉的水合物抑制性僅在一定程度內有所增強。

圖6 不同質量分數增黏劑水溶液的介觀結構Fig.6 Meso-structures of tackifier aqueous solution with different mass fraction
CMC水溶液中薄片狀和線狀水化分子聚集體的體積相對較小且分布較為均勻,網絡骨架的比表面積較大,孔隙空間尺寸較小且形態較復雜,所以親水的CMC水化分子能夠束縛更多的水分子在其附近,孔隙空間中自由水分子的數量也相對較少,致使CMC的水合物抑制性強于改性淀粉。CMC加量達到0.3%~0.5%后,水化分子聚集形成的網絡骨架變厚、變粗,表面積有所增大,孔隙空間尺寸與形態也會變得更小、更復雜(圖6),孔隙空間中可能幾乎沒有自由水分子存在,并且溶液中的水分子基本為強束縛水(水分子于溶液中很可能以強束縛水和弱束縛水的狀態存在[26],如圖7所示),進而在水合物形成驅動力較弱的情況下,水分子很難甚至無法在氫鍵作用下定向排列形成籠型結構[27-28],幾乎徹底抑制了水合物的形成,這一抑制作用與動力學抑制劑在特定條件下徹底抑制水合物形成的機制相似[29-30]。
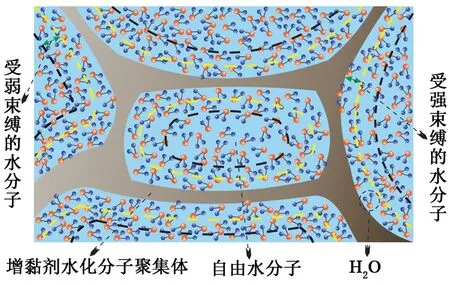
圖7 溶液中水分子的空間分布特征Fig.7 Spatial distribution characteristics of water molecules in solution

圖8 各組增黏劑水溶液的水活度Fig.8 Water activity of each solution
XC加量為0.1%和0.2%時,雖然薄片狀和細線狀水化分子聚集體比相同質量分數CMC水溶液中的體積更小、分布更均勻,而且骨架結構中的孔隙空間尺寸更小、形態更復雜,但水合物抑制性卻略弱于CMC;結合圖8中這兩個質量分數條件下溶液水活度分析得出,雖然XC水化分子聚集體在溶液中的比表面積更大,但其束縛水分子的能力相對較弱,受束縛的水分子相對較少,導致水合物抑制性略弱于CMC。XC加量為0.3%時,其水化分子在溶液中組成的骨架結構變得更粗密,孔隙空間尺寸也變得更小,雖然其水化分子束縛水分子的能力相對略弱,但在骨架結構比表面積較大的影響下,孔隙空間中的水分子很可能同樣均以強束縛水的狀態而存在,所以幾乎沒有水合物形成。XC加量增大到0.4%和0.5%后,網絡骨架結構進一步變粗、變復雜,孔隙空間進一步變小(圖6),且孔隙空間中的水分子基本為強束縛水,但溶液中卻有少量水合物形成,這很可能與XC水溶液具有較強的發泡性和穩泡性有關[31]。
研究[31]表明,改性淀粉和CMC水溶液雖然在攪拌作用下具有一定的發泡性,但由于穩泡性較差形成于溶液中的氣泡很容易破裂消散,難以穩定存在。XC分子含有大量的剛性基團以及棒狀螺旋結構,當其達到一定含量后,可在氣液界面形成穩固致密的復合膜,增大了氣泡壁的強度和厚度,使得氣泡很容易形成并且在較劇烈的動態條件下仍可穩定存在[32]。因此溶液中XC加量達到0.4%和0.5%后,在攪拌作用下溶液于反應釜中會形成大量穩定存在的甲烷氣泡,而氣泡壁處氣液接觸面積較大,極有可能少量水合物以薄膜形態形成于氣泡表面[33],從而弱化了這兩個加量條件下XC的水合物抑制性。
水合物形成驅動力較強時,被增黏劑水化分子強、弱束縛的水分子都參與到水合物的形成中,雖然無法減少水合物的形成量,但卻能夠一定程度地減緩水分子形成籠型結構,所以各組增黏劑水溶液對水合物形成的抑制作用體現在了延緩水合物形成速率上,且這一抑制作用隨增黏劑質量分數的增大逐漸增強。分析得出,此時增黏劑抑制水合物形成的能力可能主要是受水化分子附近受束縛水分子的數量所影響,并且被水化分子強束縛于表面的水分子量起到了決定性作用。雖然XC在水合物形成驅動力較弱時水合物抑制性略弱于CMC,但其水化分子聚集體的體積更小,比表面積更大,并且在溶液中組成的網絡空間結構更為復雜,更多的水分子被其強束縛于表面,促使XC減緩水合物形成速率的能力強于CMC和改性淀粉。
綜上所述,3種增黏劑水化分子在溶液中聚集形成了不同空間形態的網絡狀骨架結構,并通過約束和阻礙其附近水分子的遷移運動,在空間與時間上對骨架孔隙中水合物的成核與生長過程產生不同程度的影響,進而在不同驅動力條件下不同程度地抑制了水合物的形成。
3.2 增黏劑在不同鉆井液體系中適用性
水合物鉆井過程中為有效抑制儲層中水合物的大量分解,需盡可能保持井內溫度場平衡,即維持鉆井液處于較低的溫度條件下[9]。但低溫鉆井液在井內循環時溫壓條件處于持續變化狀態,而鉆井液的黏度在溫度變化的影響下也會不斷改變,進而影響了鉆井液的流變性[34]。所以在優選水合物鉆井液增黏劑時,不僅要考慮增黏劑對水合物形成的影響,還要考慮其提黏能力及變溫條件下鉆井液黏度的變化幅度,從而使得所配制鉆井液的流變性具有較強的可控性。考慮到海洋水合物鉆井所涉及的溫度范圍(3 ℃~室溫)[18],對5~20 ℃條件下各組增黏劑水溶液的旋轉黏度進行測試,并計算得出各組溶液在不同溫度條件下的表觀黏度和塑性黏度,結果見圖9。
分析圖9可知,各組溶液的表觀黏度與塑性黏度皆隨增黏劑加量的增大或體系溫度的降低而增大,這與增黏劑水化分子所組成的骨架結構和分子熱運動的變化密切相關,質量分數的增加會使網絡骨架變粗且形態變復雜,同時孔隙空間也會變小,減少了自由水的量,更多的水變為強與弱的束縛水,增大了剪切破壞骨架結構的難度;而體系溫度的降低會使溶液中構成網絡骨架的增黏劑水化分子與孔隙空間中水分子的熱運動減弱,且水分子對于增黏劑水化分子之間相對運動所起的緩沖作用也會變弱,從而使得增黏劑水化分子之間更加難以發生相對運動[19]。同時,受增黏劑水化分子在溶液中聚集形成網絡骨架聚合度不同的影響,CMC的提黏能力最強,XC次之,改性淀粉較弱;受溶液熱穩定性不同的影響,低溫條件下改性淀粉和XC水溶液流變性的可控性相對較強,CMC水溶液較差[35]。
綜合分析水合物形成模擬實驗及溶液黏度測試實驗的實驗結果得出,對于不需做鉆井液冷卻處理的海洋常規油氣鉆井而言,在可能出現水合物形成進而誘發安全隱患的情況下,優選鉆井液增黏劑時僅需考慮其提黏能力及水合物抑制性;如果井下溫壓條件處于水合物相平衡點附近,鉆井液中添加達到或高于0.3%的CMC后,在實現增大鉆井液黏度的同時又能有效抑制水合物的形成,鉆井液中可以少加甚至不加水合物抑制劑;當井內溫壓條件對水合物形成具有較強的驅動作用時,選擇XC作為鉆井液增黏劑能夠為含水合物抑制劑的鉆井液體系有效預防水合物形成提供更為可靠的保障。水合物鉆井施工過程中,由于需向井內泵入溫度較低的鉆井液以達到抑制儲層中水合物大量分解的目的,所以優選增黏劑時不但要考慮提黏能力和水合物抑制性,還要考慮變溫尤其是低溫條件下鉆井液流變性及流變性的可控性是否良好,所以無論井內溫壓條件對水合物形成具有或強或弱的驅動作用,XC都更為適用。
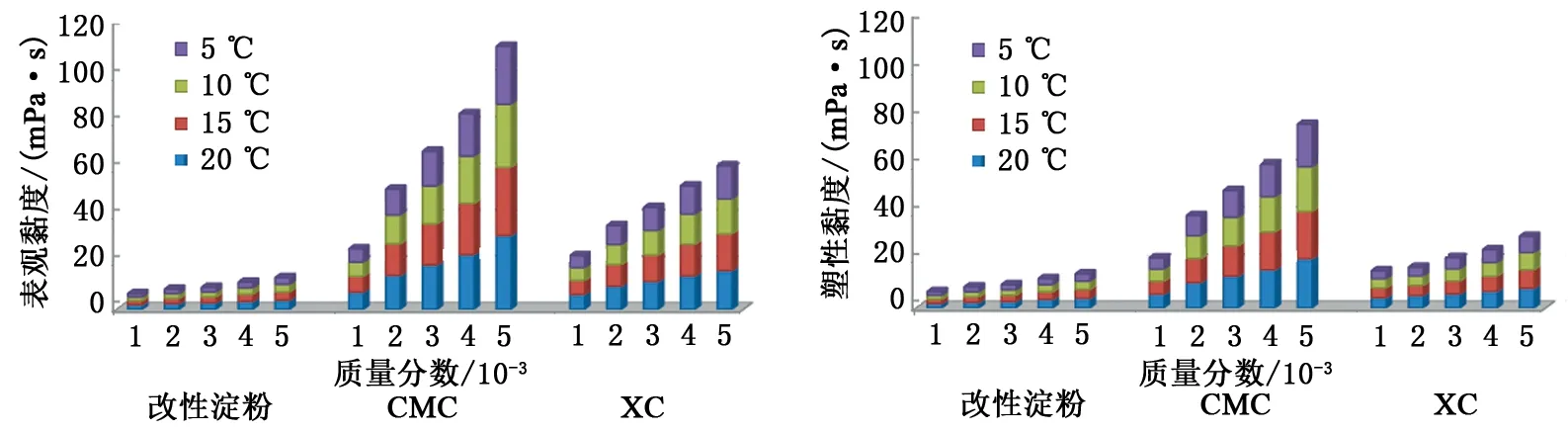
圖9 不同溫度條件下各組溶液的表觀黏度和塑性黏度Fig.9 Apparent viscosities and plastic viscosities of solutions under different temperatures
4 結 論
(1)水合物形成驅動力較弱的情況下,加量為0.1%~0.5%的改性淀粉對水合物形成具有一定抑制作用,但不能徹底抑制水合物形成,而CMC的加量為0.3%~0.5%時則可達到幾乎徹底抑制水合物形成的效果;雖然XC加量為0.3%時基本上徹底抑制了水合物的形成,但加量增大到0.4%和0.5%后,受溶液較強的發泡性和穩泡性影響,水合物抑制性又有所減弱。
(2)水合物形成驅動力較強的情況下,隨著加量的增大,3種增黏劑均能夠一定程度地減緩水合物形成速率,且由于XC分散于水溶液中可形成更加復雜的網狀空間結構,能夠束縛更多的水分子,因此其水合物抑制性強于CMC和改性淀粉。
(3)深海常規油氣及水合物鉆井過程中,若井下溫壓條件對水合物形成的驅動作用較弱,CMC更適用于不需做降溫處理的鉆井液體系,XC因其在低溫條件下流變性的可控性較強,更適用于需做降溫處理的鉆井液體系;若井下溫壓條件對水合物形成的驅動作用較強,XC則更適合用來配制需具水合物抑制性的鉆井液體系。
猜你喜歡
小獼猴智力畫刊(2022年9期)2022-11-04 02:31:42
中學生數理化·中考版(2022年11期)2022-02-16 07:01:20
小哥白尼(趣味科學)(2019年6期)2019-10-10 01:01:50
發明與創新(2016年38期)2016-08-22 03:02:52
太空探索(2016年5期)2016-07-12 15:17:55
中國塑料(2016年12期)2016-06-15 20:30:07
中國塑料(2016年5期)2016-04-16 05:25:36
中國塑料(2015年3期)2015-11-27 03:41:38
中國塑料(2015年11期)2015-10-14 01:14:14
中國塑料(2015年9期)2015-10-14 01:12:17
