UV/H2O2對化工廢水中2,6-二氯吡啶的降解研究
2021-03-05 15:48:24谷振超鄔東陳莉榮
湖南大學學報(自然科學版) 2021年2期
谷振超,鄔東,陳莉榮
(內蒙古科技大學能源與環境學院,內蒙古包頭,014010)
2,6-二氯吡啶(2,6-DCLPY)是一種應用廣泛的含氮雜環化合物,不僅是許多殺菌劑、香料和特定農醫藥物的基本原料[1],還經常作為染料、樹脂和部分精細化學品生產過程中的溶劑使用[2].因此,它普遍存在于焦化廢水、醫藥廢水和精細化工等工業廢水中[3-7].這些化工廢水中的2,6-DCLPY 因結構中含有難降解的吡啶環結構并具有較強的生物毒性,難以被一般物化處理工藝或生化手段去除[8-9].此外,它具有較強的水溶性和生物活性,可以在土壤、水體和沉積物中遷移和富集,對生態安全構成直接威脅[10].已經從包括北海(0.07 ng/L)、英國沿海(0.13 ng/L)、易北河(8.76 ng/L)等環境水體中檢測出2,6-DCLPY及多種芳香族衍生物的存在[11].因此,如何高效去除這類含吡啶環結構的污染物成為了水污染領域的研究熱點.但由于取代基團的影響,這些化合物與目前研究者們普遍關注的典型污染物之間存在一些性質上的差別.比如2,6-DCLPY 與吡啶相比,就具有堿性弱難以質子化、水溶性差難極化、環上電子密度低不易發生親電反應的特點,探明這類化合物的降解特性是對現有研究的重要補充.
目前為止,通過生物降解[8]、光化學[12]和高級氧化[13-15]等方法去除這類物質的研究均有報道,而高級氧化技術成為當前的主流研究方向[16-18].在多種生成·OH 的體系中,紫外光活化H2O2由于具備條件易控、產·OH 效率高、較低的消光系數(ε254nm=19.6 L·mol-1·cm-1)等特點而受到關注.相較于電催化、金屬離子催化等手段,通過UV 活化H2O2的過程受環境因素影響較小;無需引入新物質,可以避免催化劑分離的問題;且生成紫外光的技術已相對成熟.已有研究表明該體系可以實現系列2-鹵代吡啶的高效光解除去和礦化,對2-氯代吡啶的去除率高達95%[19].
基于此,本文選擇2,6-二氯吡啶為主要研究對象,設計了其在UV/H2O2體系下的降解實驗,研究pH、H2O2用量、底物初始濃度等因素對2,6-DCLPY降解和礦化程度的影響;考察4 種化工廢水中含量較高的陰離子在高濃度條件下對降解過程的影響;并分析了吡啶環上不同取代基對降解程度和速率的影響,以期為實際工程中采用UV/H2O2工藝去除氯代吡啶類氮化物提供參考.
1 材料與方法
1.1 試劑與儀器
實驗用水:自配2,6-二氯吡啶(2,6-DCLPY)模擬廢水,將1 g 的2,6-二氯吡啶溶解于甲醇中并定容至100 mL,常溫下保存不超過24 h.實驗時取0.2、0.4、0.6 mL 配置好的2,6-DCLPY 溶液加入到200 mL 去離子水中,得到底物質量濃度為10、20、30 mg/L 的2,6-DCLPY 溶液.
試劑與儀器:2,6-二氯吡啶(97%,阿拉丁);過氧化氫30%(分析純,天津市風船化學試劑科技有限公司);碳酸氫鈉(NaHCO3),硝酸鈉(NaNO3),氯化鈉(NaCl)和硫酸鈉(Na2SO4)(均為分析純,天津市風船化學試劑科技有限公司);精密酸度計(PHS-3C,上海雷磁儀器廠);紫外可見分光光度計(UV180G,天津光澤科技有限公司);TOC 分析儀(TOC-VCPN,日本島津公司);超純水機(GWA-UN1-20,北京普析通用儀器有限公司);COD 快速測定儀(5B-3C,連華科技);光化學反應器(ZQ-GHX-I,上海爭巧科學儀器有限公司,反應暗箱配備高壓汞燈光源,波長250~720 nm,功率300 W,工作電壓(110 ± 10)V,汞燈置于雙層全石英冷阱內,反應容積為250 mL,反應器中心光強Is=1.72 mW·cm-2).
1.2 實驗裝置與方法
UV-H2O2反應裝置如圖1 所示,采用連續反應方式.反應器總體積為2.0 L,有效反應容積250 mL,高為340 mm,外筒直徑為90 mm.石英冷阱高為375 mm,外徑60 mm,內徑40 mm.直徑為20 mm 的300 W 紫外高壓汞燈(厚1.8 mm 的石英套管保護燈管)豎直放置于內筒中央,連接循環冷卻水保證反應體系維持在室溫,反應器放置于黑箱中.首先用H2SO4與NaOH 調節水樣至所需pH 值,然后將200 mL 水樣從其中一個取樣口部倒入反應器中,投加適量30%H2O2,插入紫外燈管并固定,開啟冷凝循環水和風扇,然后接通電源開啟紫外燈和磁力攪拌器(攪拌速度80 r/min),反應開始進行.通過基礎實驗確定最優反應條件,在最優反應條件下,延長反應時間,定時取樣,測定水樣特征吸光度表征水樣中2,6-二氯吡啶的降解程度,測定TOC 值表征水中有機物的礦化程度.
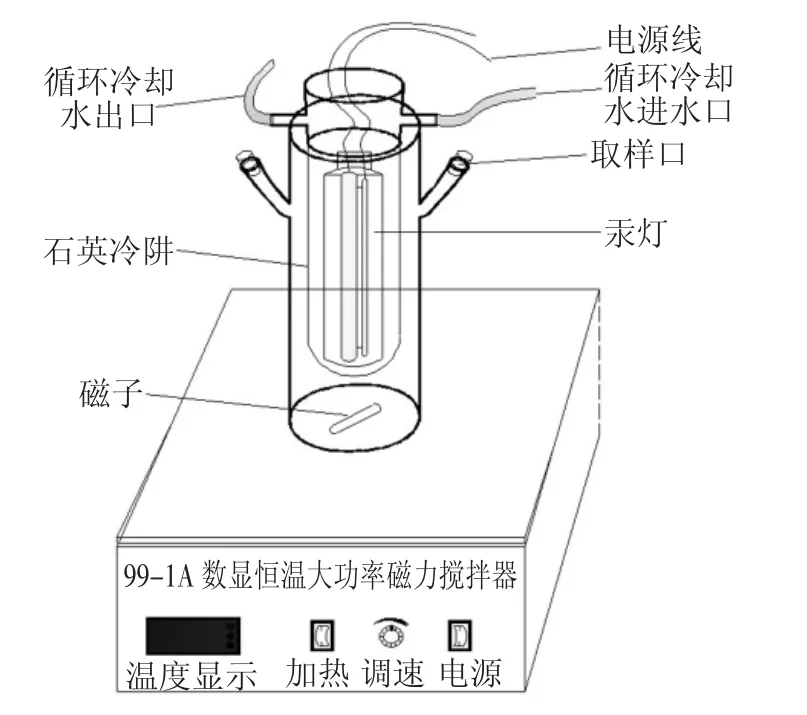
圖1 UV-H2O2反應裝置示意Fig.1 Schematic diagram of UV-H2O2reaction device
1.3 分析方法
pH 值用pH 計測量;總有機碳采用TOC 檢測儀,水樣經0.45 μm 濾膜過濾后進行TOC 的測定;采用紫外可見分光光度法(UV180G,石英比色皿為1 cm)測得2,6-二氯吡啶最大吸收峰(271 nm),然后用標準曲線法測定降解過程中2,6-二氯吡啶的質量濃度,檢測范圍為2~32 mg/L,所有測定都是將樣品調至pH 為7(±0.2)下進行;實際水樣的總堿度用國標法測定;實際水樣的COD 用連華科技COD 快速測定儀(5B-3C)測定.
2 結果與討論
2.1 2,6-二氯吡啶的降解
為了更好地評估UV/H2O2系統下2,6-DCLPY的降解效果,建立偽一級動力學模型(方程1)計算2,6-DCLPY 降解的結果.建立量子產率(φ2.6-DCLPY)模型(方程2)來計算UV 的量子產率進而評估UV 的直接光解效果.

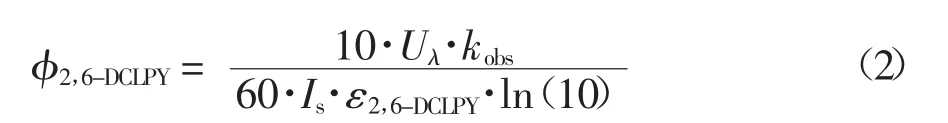
式(1)中:[2,6-DCLPY]0是2,6-二氯吡啶的初始質量濃度(mg/L),[2,6-DCLPY]是2,6-二氯吡啶在t時刻的質量濃度(mg/L),kobs是偽一級動力學反應速率常數(min-1).
式(2)中:Uλ是在特定紫外輻射波長下1 mol 光子(J Einstein-1)的能量(Uλ=hcNA/λ,得271 nm 下的單位摩爾光子能量為441 326.49 J Einstein-1),Is是平均光照強度,這里是17.2 Wm-2,ε2,6-DCLPY由朗伯-比耳定律計算(A=ε2,6-DCLPYlc).
由圖2(a)可見,當2,6-DCLPY 的初始質量濃度為10 mg/L,H2O2濃度為2.5 mmol/L,未調節配置模擬廢水的pH 值(pH 值接近中性),反應120 min后,單獨的H2O2氧化技術對2,6-DCLPY 的最大降解率僅為10%左右,反應速率常數為0.008 min-1.相同實驗條件下采用UV 系統降解2,6-DCLPY 的最大降解率為48.8%,降解速率也同步提高了5 倍.出現這樣的結果可能是因2,6-DCLPY 的最大吸光帶在271 nm,此波長下的計算φ2.6-DCLPY為0.031,這雖與之前的研究相比略低[20],但是這與高壓汞燈的主波長313 nm 較為接近,因此基本保證了用于降解2,6-DCLPY 的光量子充足,故而底物可在紫外光照射下發生直接光解.作為對照,UV/H2O2的聯合顯著提高了反應體系對2,6-DCLPY 的降解程度(最大降解率77.3%,反應速率常數0.064 min-1),這可能是由于UV/H2O2體系下2,6-DCLPY 除了直接光解的途徑外,還會被紫外光激發產生的·OH 氧化降解,見式(3)(4).

為確定是否存在·OH 參與2,6-DCLPY 降解過程,向溶液中添加對·OH 具有強淬滅作用的叔丁醇(TBA)進行對比實驗[21].在不同pH 條件下將200 mmol·L-1的TBA 加入到原反應體系進行降解實驗,發現2,6-DCLPY 的降解均被明顯抑制.圖2(b)顯示了使用淬滅劑后的實驗結果,在沒有添加任何淬滅劑的情況下最優可除去77.3%的2,6-DCLPY,而TBA 的添加使得在pH 值為3、7.2、9 的條件下2,6-DCLPY 的降解率分別降低至39.8%,32.2%和27.3%.
自由基抑制實驗表明:不含α-H 的叔丁醇在中性及弱堿性條件下對羥基自由基的清除效果明顯(kobst=3.8-7.6×108L·mol-1·s-1)[22-23].結合空白組作為對照可知,在酸性、中性和堿性條件下羥基自由基都有產生,但在中性及弱堿性條件下羥基自由基的活性更強.進一步地通過方程(1 -(kobs,TBA/kobs))來評估·OH 對2,6-DCLPY 降解的貢獻率,得出UV/H2O2體系下由·OH 氧化引起的2,6-DCLPY 去除率占總去除率的74.1%,而起協同作用的UV 只提供了25.9%的降解貢獻率.因此,·OH 是UV/H2O2體系中降解2,6-DCLPY 的主要活性物質.
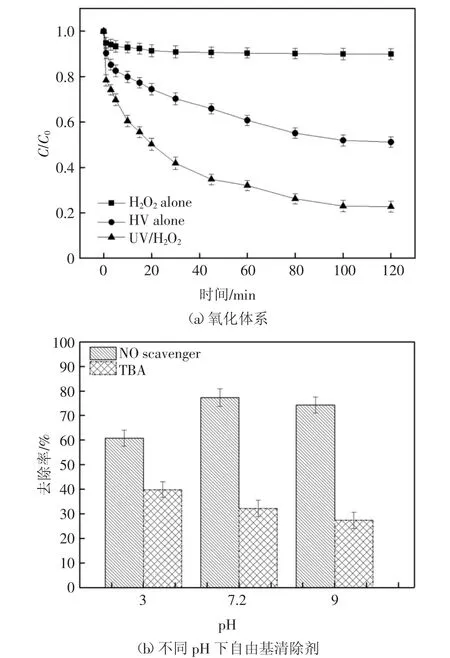
圖2 氧化體系及不同pH 下自由基清除劑對2,6-DCLPY 降解的影響Fig.2 Effect of oxidation system and scavenger at different pH on 2,6-DLPY degradation
2.2 初始H2O2濃度對降解的影響
如圖2 所示,UV 體系下H2O2的投加能夠加快2,6-DCLPY 的去除,且在一定范圍內隨H2O2濃度升高,2,6-DCLPY 的降解效率和反應速率越高.因此,UV/H2O2對2,6-DCLPY 的降解符合準一級反應動力學,不同H2O2投加量時2,6-DCLPY 反應速率常數Kobs見圖3 內插圖.當H2O2濃度小于2.5 mmol/L,2,6-DCLPY 降解速率隨著H2O2用量的增加增長較快,但當H2O2用量超過2.5 mmol/L 時,2,6-DCLPY降解速率隨著H2O2濃度的提高緩慢下降.由于H2O2可以與·OH 發生反應,見式(6),因此,過量的H2O2可能會和目標污染物競爭·OH 而影響其與底物的反應,同時高H2O2投加量下可能會存在·OH 的自清除和自復合,見式(5)(6)[24].據此確定H2O2和2,6-DCLPY 的最佳摩爾比為37 ∶1.
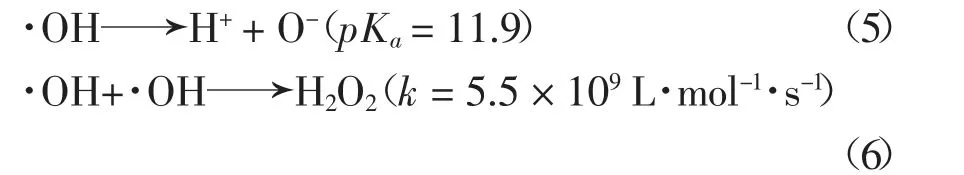

圖3 H2O2濃度對2,6-DCLPY 降解的影響Fig.3 Effect of H2O2concentration on 2,6-DCLPY degradation
2.3 2,6-二氯吡啶初始濃度對降解的影響
在過氧化氫與2,6-DCLPY 摩爾比相同的條件下,以初始質量濃度10、20、30 mg/L 的2,6-DCLPY溶液為研究對象,考察UV/H2O2體系對不同濃度2,6-DCLPY 的降解效果及礦化程度的影響.由圖4可知,2,6-DCLPY 降解率和TOC 去除率都隨反應時間延長逐漸增大.反應80 min 時,初始質量濃度10、20、30 mg/L 的溶液中2,6-DCLPY 去除率分別為73.82%、71.33%、66.77%;但溶液中有機物的礦化程度較低,在降解120 min 后TOC 去除率均低于50%.較低的礦化程度是多種原因所致,首先2,6-DCLPY 中吡啶環受到多氯取代的影響,電子密度明顯下降,增大·OH 氧化開環難度;反應前后可以觀察到pH 值的明顯變化,說明在中間過程Cl 原子轉化為氯化氫,較低的pH 會抑制后續降解過程;N 原子轉變為小分子有機胺后難以進一步礦化;隨著反應時間延長,初始添加的H2O2可能不足以提供充足的·OH.
表1 為3 種反應動力學擬合方程及參數,從中可以看出10、20、30 mg/L 時2,6-DCLPY 的反應表觀速率常數分別為1.481、1.457、1.276,2,6-DCLPY的降解速率隨初始濃度的升高而下降.整個降解過程的TOC 去除率增長緩慢,低底物質量濃度(10 mg/L)的TOC 最終去除率約為50%,高底物質量濃度(30 mg/L)的TOC 去除率僅為26.5 %,這表明大部分2,6-DCLPY 轉化為中間產物后難以進一步礦化.

圖4 底物質量濃度對2,6-DCLPY 降解的影響Fig.4 Effect of substrate concentration on 2,6-DCLPY degradation
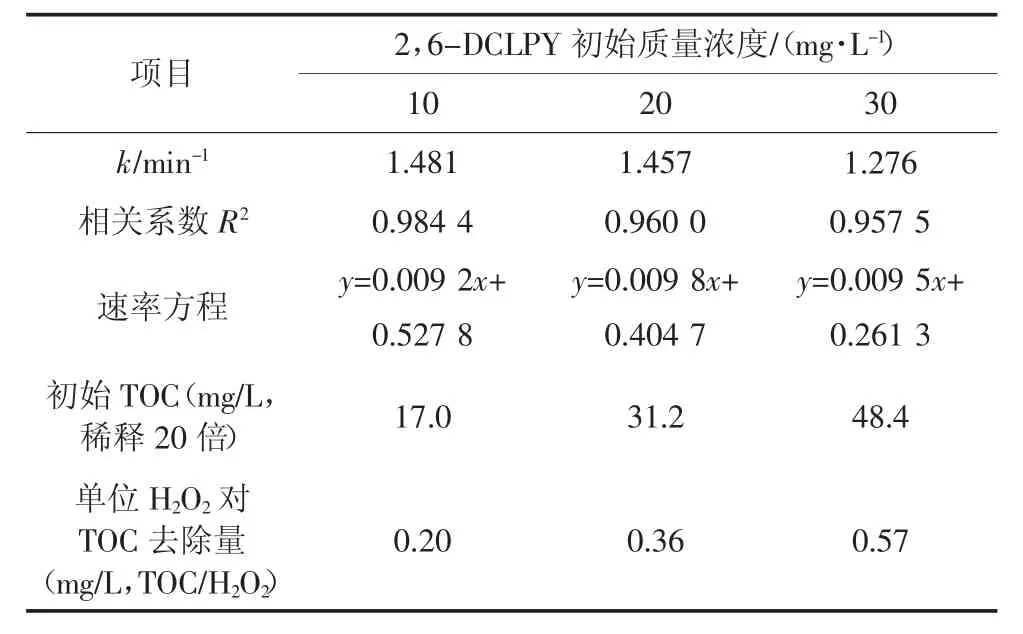
表1 UV/H2O2降解不同初始質量濃度2,6-DCLPY溶液的動力學及TOC 參數Tab.1 Kinetics and TOC parameters of different initial concentrations on 2,6-DCLPY degradation by UV/H2O2
2.4 pH 值對降解的影響
實驗前用0.1 mol/L 的HCl 和NaOH 調節體系的初始pH 值分別為3、5、7.2(2,6-DCLPY 原液pH 值為7.2,誤差為±0.02)、9、11 進行降解實驗,結果見圖5.
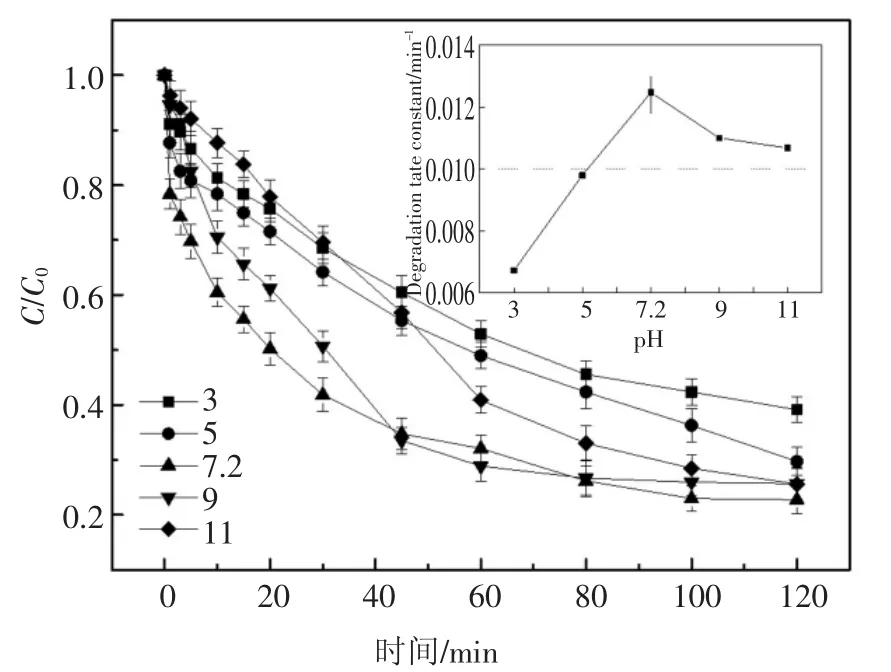
圖5 pH 值對2,6-DCLPY 降解的影響Fig.5 Influence of pH on 2,6-DCLPY degradation
結果顯示酸性條件下降解程度總體較低,酸性增強時降解率降低,但降解速度在實驗初期會出現反轉.這可能是由于弱酸性(pH=5)條件下H2O2較穩定,而較強酸性(pH=3)條件下初期高濃度的H2O2容易與·OH 反應產生HO2·,從而暫時降低了·OH 的濃度,使降解速率下降[25-26],見式(7);當溶液呈中性和弱堿性時2,6-DCLPY 降解程度較高,同時kobs和降解速率在pH 值5~9 范圍內隨pH 值提高而增加,當pH 在7~9 的范圍內時反應速率常數均在0.011 min-1以上.
考慮到降解2,6-DCLPY 的主要活性物質是·OH,而H2O2堿性電離產生的,以及OH-都會與其發生反應,見式(8)[27]、式(9)[28],從而抑制降解過程,因此理論上隨pH 上升該體系對2,6-DCLPY 的降解能力會逐漸下降,但實驗結果顯示降解率在中性(pH 7.2)時達到最高.這可能是受2,6-DCLPY 的形態變化影響,在中性及弱堿性條件下2,6-DCLPY為分子態,而較低pH 值下會結合質子變為陽離子態,此時其結構中吡啶環的電子密度降低,難以被·OH 氧化,從而導致降解率降低,實驗中堿性條件下降解反應的二級速率常數大于酸性條件也支持該解釋.
2.5 水相中無機離子的影響
化工濃鹽水中常見無機陰離子主要為Cl-,,因此設計實驗研究以上離子對2,6-DCLPY 降解過程的影響.由于Cl-和通常含量較高,是主要的鹽度組成,而,含量相對較低,因此分別選擇質量濃度范圍0~50 g/L 和0~5 g/L作為研究區間.實驗結果如圖6(a)(b)(c)(d)所示.
由圖6(a)可知,Cl-初始質量濃度從0 增加到10 g/L 時,UV/H2O2體系降解2,6-DCLPY 80 min 后去除率從73.8%下降到66.9%,而當Cl-初始質量濃度進一步增加到50 g/L 時,去除率為44.8%,與Cl-初始濃度為0 時相比,降解率下降29.0%,降解率差異并不巨大.在UV/H2O2體系下,Cl-與·OH 形成ClOH-的逆反應非常迅速,使得Cl-對·OH 的清除作用很弱.產生的ClOH-.進一步反應為Cl·和Cl2·-也有一定的氧化電勢(E0(Cl·/Cl-)=2.41 V,E0(Cl2·-/2Cl-)=2.41 V)[29],如式(10)(11)(12)中所列[29-30].因此,理論上Cl-對體系中·OH 的濃度幾乎沒有影響,不會顯著影響2,6-DCLPY 的降解.實驗中Cl-對2,6-DCLPY 降解的輕度抑制可能是由于高鹽度下參與競爭·OH 的Cl-數目巨大而阻礙了底物和·OH 的結合.

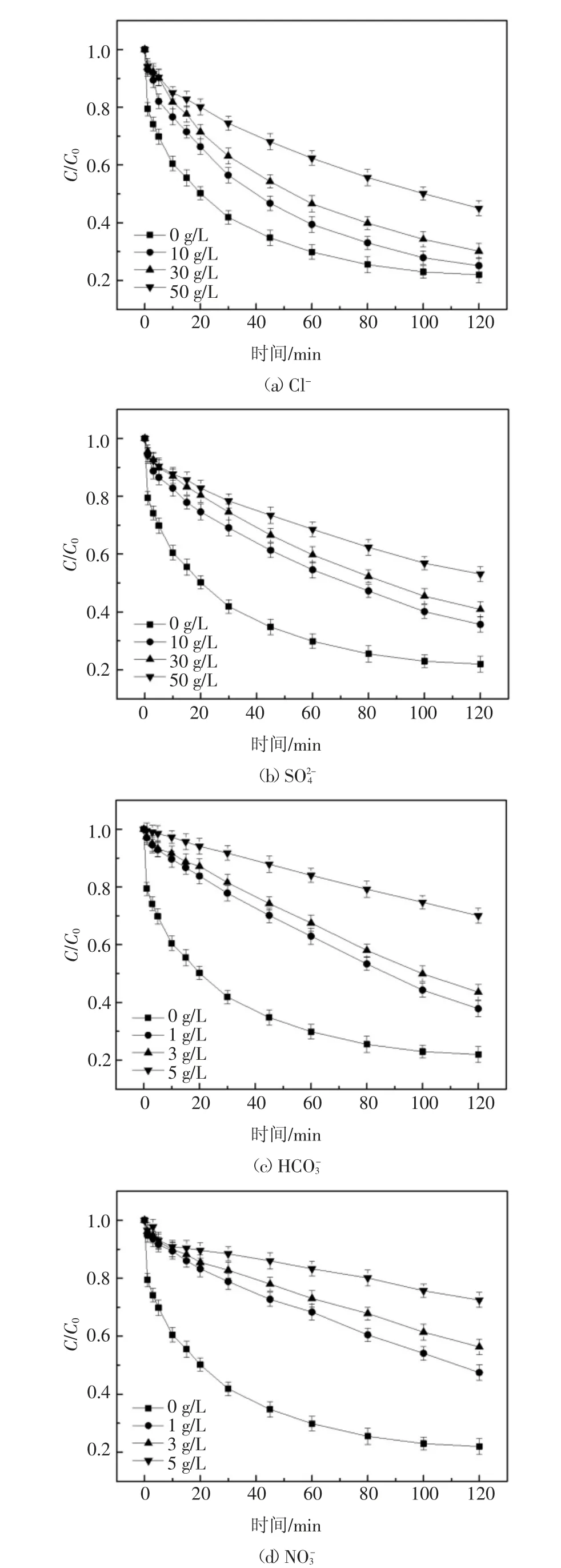
圖6 無機陰離子對2,6-DCLPY 去除的影響Fig.6 Effect of inorganic anions on 2,6-DCLPY degradation
2.6 吡啶環上不同取代基對降解效果的影響
從表2 中可以看出,吡啶環上存在強給電子基團,3-氨基-2-氯吡啶的降解速率表現為零級反應,B.F.Abramovic 等[38]對其進行了Langmuir-Hinshelwood(L-H)的動力學模型擬合分析,計算得出吸附系數為1.2×104dm3/mol.這說明反應物極為活潑,在催化劑表面吸附的瞬間即被分解,因此反應速率與反應物濃度無關,只與催化劑的反應位點數量相關.最終3-氨基-2-氯吡啶的降解率為95.0%,TOC 去除率也達90.0%.
而吡啶、2-氯吡啶、2,6-二氯吡啶在各自反應體系下均為準一級反應,三者的反應速率表現為吡啶>2-氯吡啶>2,6-二氯吡啶,這應該是由于吸電子基團Cl 的影響.根據文獻報道,吡啶環降解的主要途徑都要通過·OH 對其進行親電取代反應以生成中間產物[41],而氯取代基使得吡啶環上電子密度下降,降低了吡啶環的反應活性,抑制了降解的進行,氯取代基團越多則該影響越顯著.因此吡啶環上電子密度最低的2,6-二氯吡啶降解和礦化程度最低.
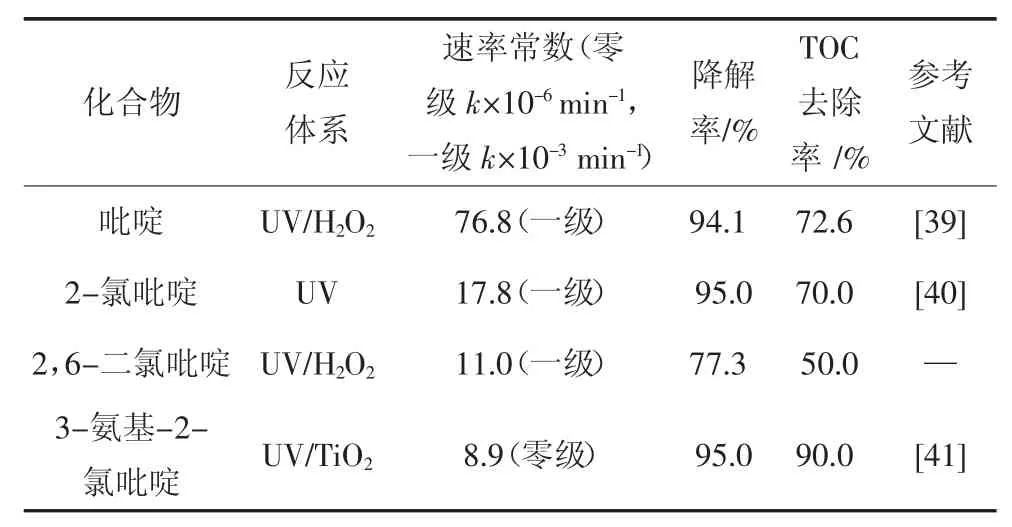
表2 不同取代吡啶的降解效果Tab.2 Degradation effect of different substituted pyridines
2.7 2,6-二氯吡啶在實際水體中的降解
為研究UV/H2O2對實際水體中2,6-DCLPY 的去除效果,取包頭某焦化企業的RO 濃水作實際水樣(水質參數如表3),向其中加入10 mg/L 2,6-DCLPY 和3.5 mmol/L 的H2O2進行降解實驗.實驗結果如圖7 所示,與純水中2,6-DCLPY 的降解相比,2,6-DCLPY 在實際水體中的降解受到抑制.考慮到所取實際廢水含鹽量高,其中主要陰離子Cl-和都會抑制降解過程,如上述討論,這一實驗結果符合預期.

表3 實際水樣的水質參數Tab.3 Water quality parameters of actual water samples
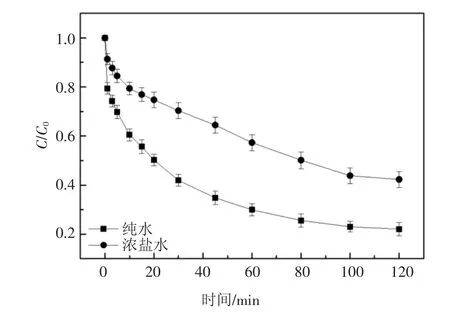
圖7 2,6-二氯吡啶在實際水體中的降解Fig.7 Degradation of 2,6-dichloropyridine in actual water
3 結論
1)·OH 是UV/H2O2體系中導致2,6-DCLPY 降解的主要活性物質,由其氧化引起的2,6-DCLPY降解貢獻了總降解量的74.1 %,而UV 引發的直接光解占另外25.9 %.2,6-DCLPY 的降解過程符合準一級反應動力學方程,2,6-DCLPY 的降解率隨H2O2用量的增加而逐漸提高,但是過量的H2O2能夠成為·OH 的淬滅劑.
2)實驗確定過氧化氫和2,6-DCLPY 的最佳摩爾比為37 ∶1,pH 為7.2 時降解效率達到最高.不同底物濃度的TOC 去除率測定實驗表明,大部分2,6-DCLPY 轉化為中間產物后難以進一步礦化.
3)UV/H2O2體系中,Cl-和的存在對2,6-DCLPY 的降解均有抑制,其離子濃度越高抑制作用越明顯和的抑制效果更加顯著,因此2,6-DCLPY 在實際水體中的降解過程相比于純水中會受到一定程度的抑制.
4)給電子基團如氨基有利于吡啶類污染物的降解,吸電子基團如氯會抑制吡啶類污染物的降解,因此在吡啶、2-氯吡啶、2,6-二氯吡啶和3-氨基-2-氯吡啶四種物質中,電子密度最低的2,6-二氯吡啶最難降解和礦化.
猜你喜歡
小獼猴智力畫刊(2022年9期)2022-11-04 02:31:42
中學生數理化·八年級物理人教版(2022年3期)2022-03-16 05:55:08
當代陜西(2021年2期)2021-03-29 07:41:24
新世紀智能(數學備考)(2020年11期)2021-01-04 00:38:16
小哥白尼(趣味科學)(2019年6期)2019-10-10 01:01:50
發明與創新(2016年38期)2016-08-22 03:02:52
太空探索(2016年5期)2016-07-12 15:17:55
中國塑料(2016年3期)2016-06-15 20:30:00
新高考·高一物理(2014年1期)2014-09-18 01:26:07
中國火炬(2010年7期)2010-07-25 10:26:09
