鐵改性火山巖顆粒對水中低濃度磷吸附性能的研究
2021-03-09 01:23:48趙明杰李思晨劉曉靜栗勇田
環境科技 2021年1期
趙明杰, 李思晨, 劉曉靜, 栗勇田
(1.天津大學環境科學與工程學院,天津 300072;2.秦皇島天大環保研究院有限公司,河北 秦皇島 066000;3.河北省河道水質凈化及生態修復重點實驗室,河北 秦皇島 066000;4.秦皇島市水污染監測及治理工程技術研究中心,河北 秦皇島 066000)
0 引言
磷是生物生長所必需的元素,同時也是水體營養化的重要限制因子[1],控制磷等營養性物質進入水體是解決富營養化問題的根本途徑[2]。城鎮污水廠處理對象為生活污水和預處理后的工業廢水,經二級處理后,污水中的TP 質量濃度通常在2 mg/L 以下。隨著污水廠出水排放標準的不斷提高,污水廠出水的TP 濃度需要進一步降低,針對低濃度含磷廢水的深度處理,吸附法因其操作方便,費用低廉、綠色環保、可重復利用等優點而被廣泛應用。目前關于吸附法的研究多集中于粉狀吸附材料[3],吸附劑回收困難,容易導致吸附材料流失,限制了吸附除磷技術的應用。此外,單一的硅藻土、活性炭等吸附材料,吸附容量受限,被吸附的磷存在脫逸現象,很難達到理想的除磷效果。
火山巖作為一種儲量極大的天然礦石資源,表面疏松多孔,通常用來吸附水體中的重金屬[4-6],鐵鹽作為常見的除磷混凝劑,具有穩定高效的除磷效果[7]。本研究采用FeCl3對天然火山巖進行改性,制得改性火山巖顆粒吸附劑,并用其對低濃度含磷廢水進行吸附影響因素和吸附特性研究,以期為開發新型吸附材料處理含磷廢水提供參考。
1 實驗部分
1.1 材料與設備
天然火山巖顆粒,KH2PO4,FeCl3,硫酸亞鐵,鉬酸銨,氫氧化鈉,酒石酸鉀鈉,硫酸,鹽酸,過硫酸鉀,抗壞血酸,均為分析純;多參數水質分析儀(DGB-401,雷磁);數顯恒溫磁力攪拌器(HJ-6B,常州金壇晨陽電子儀器廠);X 射線熒光儀 (Supermini200,日本理學株式會社);X 射線衍射儀(Smartlab9,日本理學株式會社);掃描電子顯微鏡(Maia3,泰思肯貿易有限公司);電熱恒溫鼓風干燥箱(DHG-9023A,上海篤特科學儀器有限公司);pH 計(pHS-25 型,上海精密科學儀器有限公司)。
1.2 改性火山巖顆粒的制備及表征
將天然火山巖分別過3 和5 mm 篩,控制粒徑在3 ~5 mm 之間,用純水洗凈,在105 ℃下烘干,冷卻后按1 ∶5 的固液比,將火山巖顆粒置于質量分數為7%的鹽酸溶液中浸泡8 h。 用純水洗凈烘干,冷卻后按1 ∶5 的固液比,將酸改性后的火山巖顆粒置于17%的FeCl3溶液中浸泡8 h 后,取出置于200 ℃的馬弗爐中焙燒2 h,冷卻后得到鐵改性火山巖顆粒。 通過X 射線熒光儀(XRF)分析確定改性前后的元素組成,X 射線衍射儀(XRD)分析改性前后火山巖的晶體結構變化,掃描電子顯微鏡(SEM)觀察改性前后火山巖表面形貌特征。
1.3 磷吸附影響因素實驗
分別取一定量的改性火山巖顆粒加入100 mL由KH2PO4配置的模擬低濃度含磷廢水中,置于磁力攪拌器上攪拌一定時間后取出,離心分離,取上清液測定溶液中的TP 濃度,并計算TP 去除率和單位質量吸附劑的磷吸附量。 設置不同吸附劑投加量、溶液pH 值和溫度條件,同時嚴格控制其他條件不變,以確定吸附的最佳條件。 TP 濃度分析方法采用鉬酸銨分光光度法 (GB 11893—89),通過繪制TP濃度與吸光度的標準曲線確定TP 濃度,為保證數據的可靠性,每組實驗均設重復實驗,測量結果取平均值。
TP 去除率(η)和單位質量吸附劑吸附量(Qe)的計算方法分別見式(1)和式(2):
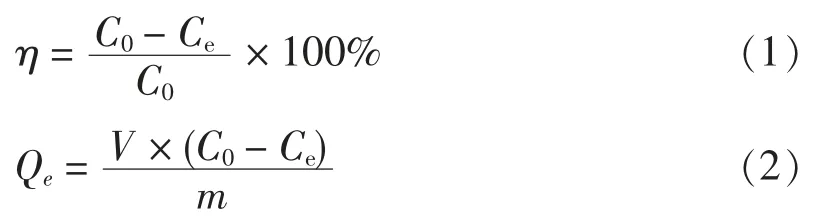
式中:η 為TP 去除率,%;Qe為單位質量吸附劑對TP 的吸附質量濃度,mg/L;V 為處理污水體積,L;C0為廢水中TP 的初始質量濃度,mg/L;Ce為吸附后TP的剩余質量濃度,mg/L;m 為吸附劑的質量,g。
2 結果與討論
2.1 改性火山巖的化學元素組成
利用XRF 對天然火山巖和改性火山巖樣品中的化學元素種類及含量進行了分析,結果見表1。

表1 火山巖改性前后的元素組成
由表1 可以看出,改性前后,火山巖的主要化學成分均以Si,Fe,Ca,K,Na 為主。 與天然火山巖相比,改性后的火山巖晶格中Ca,Na,K 等元素含量降低,這說明在鹽酸浸漬處理過程中,有效去除了火山巖中的部分金屬陽離子氧化物雜質。此外,改性火山巖中ω(Fe)增加了3.50%,ω(Cl)增加了2.11%,表明鐵鹽可能附著于火山巖表面。
2.2 改性火山巖的XRD 分析
利用XRD 對天然火山巖和改性火山巖進行分析,得到結果見圖1。

圖1 火山巖改性前后的XRD
由圖1 可以看出,火山巖中含有以Si,Al,Ca,Fe,O 元素形成的多種化合物,主要為SiO2。 與天然火山巖相比,經過焙燒后的改性火山巖主要特征峰沒有發生顯著變化,但Fe2O3峰形更加尖銳,結晶度更好,說明附著在火山巖表面的部分FeCl3轉化成了Fe2O3,火山巖層間結構沒有發生顯著的變化。
2.3 改性火山巖的SEM 分析
利用SEM 分別對天然火山巖及鐵改性火山巖樣品進行分析,結果見圖2。
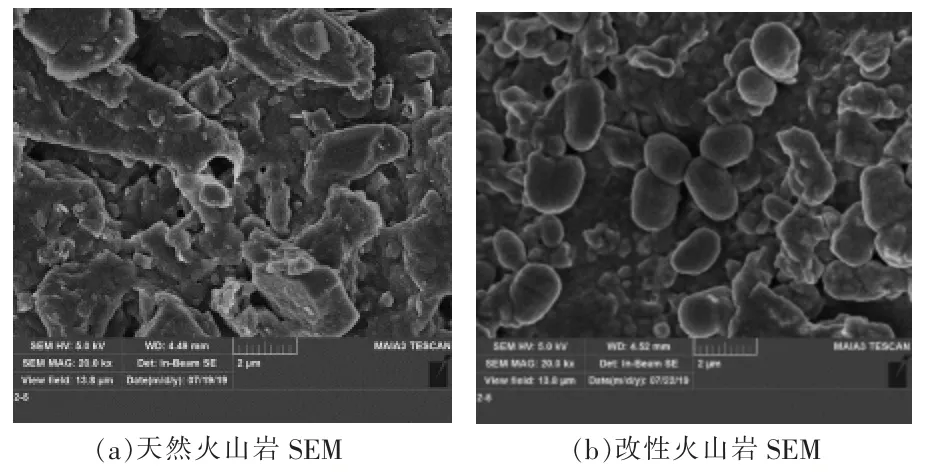
圖2 火山巖改性前后的SEM
由圖2(a)可以看出,天然火山巖表面粗糙,層狀結構較為明顯,具有較多的孔隙。 從圖2(b)可以看出,經過改性后,火山巖外層附著了大量的橢球體顆粒。 結合相關文獻[8-9]及XRD 分析結果,得知改性火山巖表面的附著物可能為α-Fe2O3,這些Fe2O3粘連堆疊在一起,一方面增大了火山巖的比表面積,另一方面也增強了火山巖的化學吸附能力。
2.4 改性火山巖對水中磷吸附的影響因素研究
2.4.1 改性火山巖投加量對磷吸附效果的影響
對水溫20 ℃,pH 值=7,TP 質量濃度為2.0 mg/L的模擬廢水進行不同投加量實驗,吸附時間為12 h,研究鐵改性火山巖吸附劑投加量對水中TP 去除率的影響,結果見圖3。
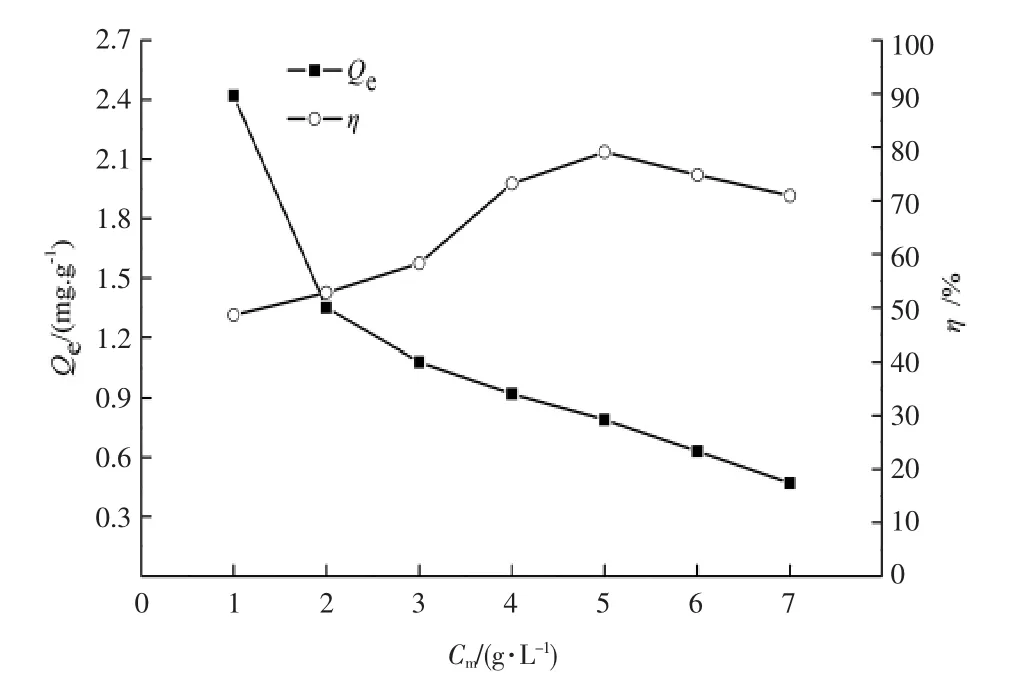
圖3 不同投加量對改性火山巖吸附除磷效果的影響
由圖3 可以看出,隨著投加量的增大,改性火山巖對水中TP 的去除率呈現先增大后略微降低的趨勢。這是因為投加量過小時,水中磷酸根離子遠多于改性火山巖能夠提供的吸附點位,剩余TP 濃度較高,隨著投加量增大,吸附劑提供的表面吸附點位隨之增多,吸附面積增大,使水中更多的磷可以附著在改性火山巖上,所以TP 的去除率升高,當投加質量濃度為5.0 g/L 時,TP 去除率最高達到了79.1%,繼續加大吸附劑的投加量,TP 去除率變化較小,過多的吸附劑可能會導致磷酸根從液相向火山巖固相表面的遷移能力降低,阻礙吸附過程的進行。 另外,吸附劑投加量的增大,會導致吸附點位發生相互掩蔽現象[10],存在空余吸附點位,致使單位質量吸附劑的吸附量下降。
2.4.2 溶液pH 值對磷吸附效果的影響
其它試驗條件一定時,控制溶液初始pH 值進行改性火山巖對水中磷的吸附實驗,結果見圖4。
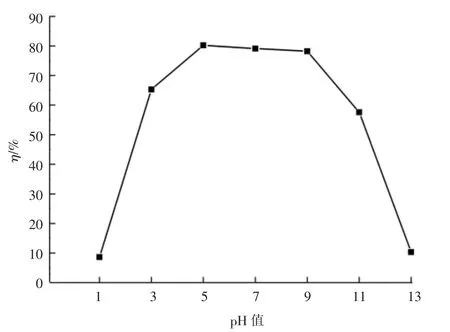
圖4 不同pH 值對改性火山巖吸附除磷效果的影響
由圖4 可以看出,隨著溶液pH 值的增加,TP 的去除率呈現先增大后趨于平緩再減小的趨勢,在強酸或強堿條件下,改性火山巖吸附除磷效果較差。這是因為磷酸的一級酸度系數為2.12,當pH 值<2.12時,水中的磷以H3PO4的形式存在,難與Fe3+結合吸附。 隨著溶液pH 值的增大,水中PO43-增多,在吸附過程中,HPO42-,H2PO4-與Fe3+結合生成絡合物,吸附反應逐漸活躍。 當pH 值為5 ~9 范圍內,TP 去除率均達到了75%以上。 而當溶液pH 值過高時,OH-濃度增加導致Fe3+濃度減少,在濃度積一定的情況下,PO43-濃度相應增大,導致除磷效果下降。
2.4.3 溫度對磷吸附效果的影響
控制其它試驗條件一定,改變溶液溫度進行改性火山巖對水中磷的吸附實驗,結果見圖5。

圖5 不同溫度對改性火山巖吸附除磷效果的影響
由圖5 可以看出,隨著溶液溫度升高,TP 去除率逐漸提升,反映出改性火山巖對水中磷的吸附為吸熱反應。這是由于溫度的升高,離子之間的碰撞逐漸增多,熱運動加劇,與吸附劑之間的碰撞增多,有利于吸附的進行。 在溶液溫度為30 ℃時,達到吸附平衡后,TP 去除率為81.8%,此時溶液中剩余TP 質量濃度為0.364 mg/L,達到GB 18918—2002《城鎮污水處理廠污染物排放標準》一級A 排放標準。 當溫度超過30 ℃后,溶液中TP 去除率基本平穩,同時考慮到實際情況,確定30 ℃為最佳溫度。
2.5 吸附動力學研究
動力學模型是對于吸附劑吸附速率的描述。 室溫下,當溶液中TP 質量濃度為2 mg/L,pH 值在7 左右,改性火山巖投加質量濃度為5 g/L 時,實測改性火山巖在不同吸附時間下的吸附量,并分別采用非線性擬一級動力學方程(式3)、擬二級動力學方程(式4)和Webber-Morris 方程(式5)對實驗數據進行擬合,見圖6,相關擬合參數數值見表2。
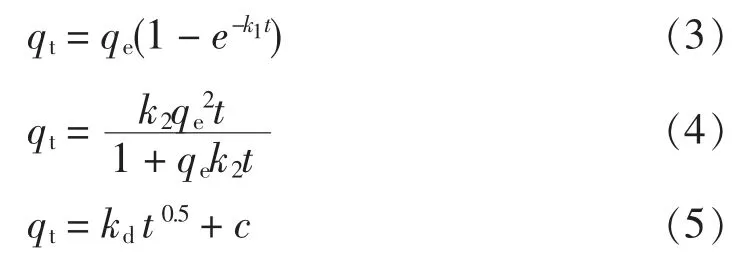
式中:t 為吸附時間,h;k1為擬一級動力學模型常數;k2為擬二級動力學模型常數;kd為Webber-Morris模型常數;qt為t 時刻的吸附質量分數,mg/g;qe為吸附平衡時的吸附質量分數,mg/g。

圖6 不同動力學模型擬合
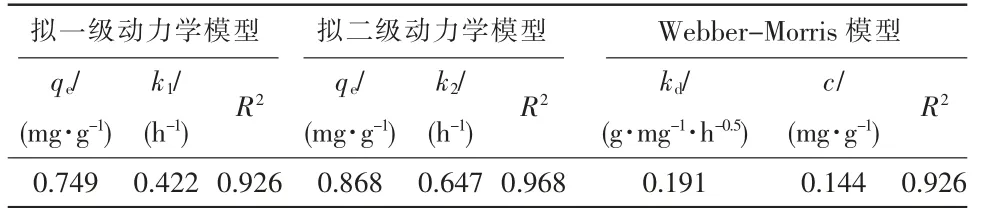
表2 不同動力學模型擬合參數
由圖6(a)可以看出,改性火山巖在初期對TP的吸附速率較快,曲線斜率較大,之后趨于平緩。0 ~12 h 中,隨著吸附時間的增加,吸附量也在逐漸增加,第12 h 時,對TP 的吸附質量分數達到0.80 mg/g,此后,吸附量趨于飽和穩定。 吸附過程由物理吸附、化學吸附和顆粒內擴散共同控制,由表2 中動力學擬合參數可以看出,擬二級動力學模型中R2為0.968,且所得平衡吸附量與實測值接近,說明反應更符合二級動力學模型。 由圖6(b)可以看出,擬合曲線沒有經過原點,說明除了顆粒內擴散外,還有其他過程對改性火山巖吸附除磷的反應速率進行控制[11]。在顆粒內擴散的過程中,火山巖的吸附共經歷3 個階段: 第一階段水中磷酸鹽擴散至改性火山巖外表面階段,這是由于吸附劑表面存在大量的吸附點位,具有較快吸附速率;第二階段水中的磷酸鹽在火山巖顆粒內部擴散階段,受到改性火山巖和磷酸鹽之間的電子共用和轉移作用,擴散阻力增大,第三階段隨著磷酸鹽濃度的降低,顆粒內部擴散逐漸減弱,直至吸附達到平衡的階段。
2.6 等溫吸附研究
當溫度一定時,吸附劑的吸附量隨著平衡濃度變化而變化的曲線稱為等溫吸附曲線,作為吸附劑吸附性能的重要參考依據。 在25 ℃室溫下,改性火山巖投加質量濃度為5 g/L,pH 值=7 時,吸附14 h后,水體水中TP 濃度基本不變,達到吸附平衡狀態,此時測量不同磷吸附平衡濃度下的吸附數據,采用Laungmuir 模型(式6)和Freundlich(式7)對實驗數據進行擬合見圖7,擬合參數數值見表3。

式中:Qe為單位質量吸附劑的吸附質量濃度,mg/L;Qm為吸附平衡時,單位質量吸附劑的最大吸附質量濃度,mg/L;KL為Laungmuir 吸附常數;KF為Freundlich吸附常數;Ce為吸附平衡時溶液中的TP 質量濃度,mg/L;1/n 為特征常數。

圖7 等溫吸附曲線模型擬合

表3 等溫吸附曲線模型擬合參數
由圖7 可以看出,隨著TP 吸附平衡濃度的增大,改性火山巖的吸附量先迅速增大,后趨于平緩,這是因為隨著污水中磷濃度的增大,水中磷酸根離子濃度與吸附劑表面液膜的濃度差增加,有利于磷酸根向吸附劑表面遷移[12],最終達到單位質量吸附劑的最大吸附量。通過表3 中擬合參數可以看出,改性火山巖吸附劑對水體中TP 的吸附行為更符合Laungmuir 等溫吸附模型,說明吸附過程主要是單分子層吸附[13],利用分離因數RL進一步分析Langmuir吸附模型,其值可依照(式8)計算。

式中:C0為初始TP 質量濃度,mg/L。
根據RL的不同數值,可將吸附分為4 類:RL=1時,吸附過程為可逆吸附;RL=0 時,吸附過程為不可逆吸附;0 <RL<1,吸附過程為優勢吸附;RL>1時,吸附過程為非優勢吸附。 在實驗濃度范圍內,Langmuir 等溫吸附曲線中,RL始終處于0 ~1 的范圍中,說明改性火山巖對水體中磷為優勢吸附。
3 結論
(1)通過FeCl3對天然火山巖進行改性,制備鐵改性火山巖顆粒吸附劑,利用XRF,XRD,SEM 對改性前后火山巖進行表征,顯示鐵有效附著于火山巖表面,增大了吸附劑比表面積及吸附點位,火山巖晶體結構沒有發生顯著的變化。
(2)在溫度為30 ℃,初始pH 值為7,TP 質量濃度為2 mg/L 的模擬污水中投加質量濃度為5 g/L 改性火山巖顆粒吸附劑,吸附12 h 后達到吸附平衡,此時水中TP 的去除率為81.8%,溶液中剩余TP 質量濃度為0.364 mg/L,達到GB 18918—2002《城鎮污水處理廠污染物排放標準》一級A 排放標準。
(3)鐵改性火山巖對水中低濃度磷的吸附為單分子層吸附,吸附速度較快,屬于優勢吸附,并由物理吸附、化學吸附和顆粒內擴散共同控制,其吸附過程更符合擬二級動力學模型及Langmuir 等溫吸附模型。
猜你喜歡
中學生數理化·八年級物理人教版(2021年12期)2021-12-31 03:23:08
中學生數理化·中考版(2020年10期)2020-11-27 01:59:48
中國生殖健康(2019年2期)2019-08-23 08:12:08
產品可靠性報告(2017年7期)2017-09-05 09:49:12
中國塑料(2016年12期)2016-06-15 20:30:07
中國塑料(2016年5期)2016-04-16 05:25:36
汽車觀察(2016年3期)2016-02-28 13:16:26
中國塑料(2015年3期)2015-11-27 03:41:38
中國塑料(2015年11期)2015-10-14 01:14:14
中國塑料(2015年9期)2015-10-14 01:12:17
