IL2RG變異致重癥聯合免疫缺陷病的臨床表現和基因分析
2021-03-16 10:09:10王高偉肖夢君謝振華李東曉羅淑穎張耀東王美燁
山西醫科大學學報 2021年2期
王高偉,劉 磊,肖夢君,謝振華,劉 菁,李東曉,羅淑穎,張耀東,王美燁
(鄭州大學附屬兒童醫院,河南省兒童醫院,鄭州兒童醫院,河南省兒童遺傳代謝性疾病重點實驗室,鄭州 450053;*通訊作者,E-mail:1005164555@qq.com)
重癥聯合免疫缺陷病(severe combined immunodeficiency,SCID)是聯合免疫缺陷病中最嚴重的類型,由多個基因缺陷引起。研究發現SCID的發生至少與以下基因相關:IL2RG、ARTEMIS、RAG1、RAG2、ADA、CD45、JAK3和IL7R[1],其中由白細胞介素-2受體共同γ鏈(interleukin 2 receptor subunit gamma,IL2RG)基因變異引起的X-連鎖重癥聯合免疫缺陷(X-SCID)占50%-60%[2]。X-SCID患者免疫系統常表現為T細胞(CD3+)、NK細胞(CD16+/CD56+)缺失或顯著減少,B細胞(CD19+)數量正常或增加,但功能異常,從而導致免疫球蛋白產生異常[3]。IL2RG編碼的IL-2Rγ鏈是IL-2、IL-4、IL-7、IL-9、IL-15、IL-21等多種細胞因子受體共有的功能組成單位,即共有鏈,是聯系細胞膜表面細胞因子結合區域與下游細胞信號轉導分子之間的紐帶。在X-SCID的臨床診斷中,T-B+NK-這種臨床表現是由IL-4、IL-7、IL-15、IL-21四種細胞因子的缺失所致,IL-7、IL-15兩種細胞因子是T和NK細胞生長所必需的,IL-4和IL-21兩種細胞因子的信號轉導紊亂會導致內在的B細胞缺陷[4-6]。IL-7在T細胞的發育中具有不可缺少的作用,其誘導的信號缺陷是X-SCID中T淋巴細胞減少的主要原因[7]。因此,IL-2Rγ鏈的完整性對于機體免疫功能至關重要[8]。根據現有研究,較早的進行基因診斷,明確病因,并及時進行骨髓或干細胞移植對本病具有一定的效果,并能延長生存期[9,10]。本文回顧1例IL2RG變異引起的X-連鎖重癥聯合免疫缺陷病,以提高對X-SCID的認識。
1 病例報告
現病史:先證者,男,3月14天,以“間斷感染14 d”來診,主要表現3月齡開始出現腹瀉,治療效果欠佳。
個人史與既往史:無特殊。
家族史:父母體健,非近親婚配。哥哥,12歲,體健。母孕期體健。患兒三舅于生后1個月夭折,死因不明。
體格檢查:精神萎靡,雙下肢、足部輕度浮腫。
實驗室檢查:血常規:白細胞(WBC)32.10×109/L,紅細胞3.9×1012/L,血小板519×109/L,血紅蛋白93 g/L,中性粒細胞百分比94.6%,淋巴細胞百分比54.8%;降鈣素原0.161 ng/ml。肝功能:谷丙轉氨酶304.8 U/L(0-40 U/L),谷草轉氨酶430.0 U/L(0-40 U/L),總膽汁酸7.1 μmol/L。免疫:患兒IgG 1.3 g/L,IgA<0.03 g/L,IgM 0.08 g/L,C3 0.99 g/L,C4 0.39 g/L,CD3+CD4+T淋巴細胞0,CD3+CD8+T淋巴細胞1%。甲胎蛋白707.2 ng/ml(0-140 ng/ml),輪狀病毒抗體陰性,感染四項、甲軸五項、呼吸道病原學、結明三項、細菌內毒素、凝血功能等均無明顯異常。腹部彩超示肝大(肋下25.5 mm)。診斷:“肝損害、免疫缺陷”?
治療:入院后給予保肝、抗感染、營養支持、補液等治療,入院第3天出現發熱、抽搐,加用頭孢曲松抗感染,人免疫球蛋白抗體治療,體溫好轉,仍腹瀉、精神差,家長要求出院,出院第12天死亡(具體不詳)。
基因分析:本研究經醫院醫學倫理委員會批準并獲得患兒父母知情同意。抽取患兒的外周血標本進行DNA提取,應用二代測序技術進行全外顯子組測序,應用Sanger測序進行位點驗證。檢索內部數據庫、dbSNP、ExAC等人群數據庫,標注核苷酸多態性和低頻良性變異。檢索HGMD,Pubmed,Clinvar等數據庫,整理相關文獻,參考ACMG(美國醫學遺傳學和基因組學學會)變異分類指南,對變異進行分類。
檢測結果顯示患兒IL2RG(NM_000206)存在半合子錯義變異c.812G>A,p.(Gly271Glu)(見圖1),位于第6外顯子上。根據美國ACMG變異分類指南,此變異符合PS1+PM2,為可能致病變異。檢測結果顯示此變異遺傳自表型正常的母親。患兒哥哥、外公、外婆、大舅、二舅在該位點為野生型。先證者家系圖見圖2。
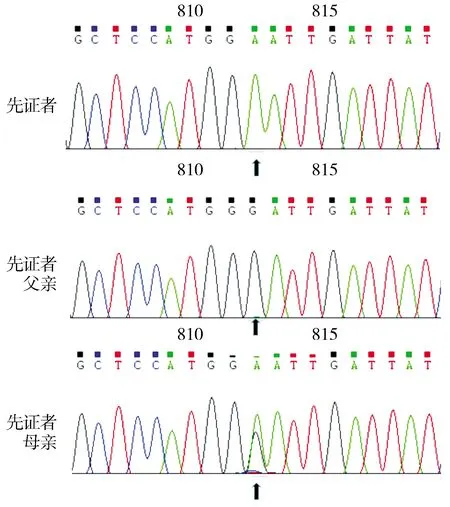
圖1 先證者及其父母IL2RG測序圖
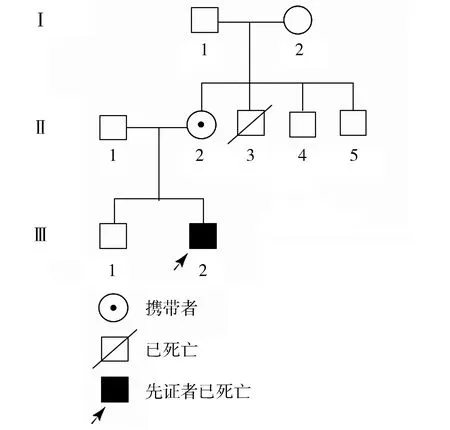
圖2 先證者家系遺傳圖
2 討論
X-SCID為SCID中最常見的一種,1993年Noguchi等[11]和Puck等[12]將X-SCID致病基因定位于X染色體q13.1上的IL2RG,其全長4 145個堿基,含有8個外顯子,編碼369個氨基酸的IL-2Rγ鏈蛋白,該蛋白對調控T細胞、NK細胞和B細胞的分化、發育和成熟起關鍵作用。IL2RG變異會導致其編碼的IL-2Rγ鏈蛋白功能障礙,從而導致T細胞發育過程中細胞內信號傳導障礙,引起T細胞功能障礙,從而導致嚴重的T細胞缺陷,進一步造成B細胞功能障礙,臨床表現為細胞免疫和體液免疫功能下降誘發機體反復感染[13]。對于多數患者而言,通常表現為肺炎、腹瀉、發熱等癥狀。
據Clinvar、HGMD數據庫顯示,目前共收錄IL2RG變異267種,突變類型包括:錯義突變、移碼突變、無義突變和剪接位點突變,以錯義突變為主。國外報道突變多發生在第3,4,5外顯子上[14]。Niemela等[15]報道了人類中37個IL2RG變異引起的X-連鎖重癥聯合免疫缺陷病,包含本文變異位點c.812G>A,p.(Gly271Glu)。本家系中先證者母親為雜合攜帶者,且表型正常,符合X連鎖隱性遺傳特征。根據美國ACMG變異分類指南,此變異符合PS1+PM2,為可能致病變異。本患兒在臨床癥狀上主要表現為嚴重的腹瀉,T細胞數量接近于0,B細胞數量雖在正常范圍之內,但各種免疫球蛋白、補體均有降低,提示B細胞功能存在異常。免疫功能幾乎喪失,符合免疫缺陷病的特點,結合基因檢測結果確定為IL2RG變異導致的X-連鎖重癥聯合免疫缺陷病。
X-SCID在臨床診斷中通過流式細胞儀測定T淋巴細胞、B淋巴細胞、NK細胞的數量具有較高的參考價值。一般情況下,母乳喂養的患兒會攜帶有母體中的抗體,能抵抗感染,存活一定時期,不易表現出臨床癥狀,因此在前期診斷中,運用分子生物學手段及早檢測極為重要。由于免疫細胞的缺失,患兒早期起病會經歷真菌、細菌感染,為治療帶來很大麻煩。在臨床診斷過程中,基因分析能很好地明確病因,并進行治療。在基因治療的研究方面,已經取得很大進展,Takafumi等[16]運用腺相關病毒作為基因載體在X-SCID小鼠骨髓細胞中的內源性的IL2RG位點插入一部分IL2RGcDNA,之后把這些體外編輯的骨髓細胞重新植入X-SCID小鼠體內,可產生CD4+和CD8+T細胞,為臨床治療提供了很好的依據。在骨髓移植治療方面,Jamal等[17]研究發現,靶向基因組編輯可在人源的X-SCID多能干細胞疾病模型中恢復T細胞分化功能。隨著臨床檢測技術和治療方法的不斷發展,早發現、早進行骨髓造血干細胞移植,可使X-SCID成活率高達95%[18]。有研究表明:當B細胞功能完整時,通過骨髓移植對患者進行T細胞的功能重建,可以使患者存活長達40年[19]。基因治療技術和相關領域分子生物學研究的不斷突破,為X-SCID患兒的治療提供更好的依據。本文通過對患兒的臨床診斷、基因檢測和家系分析,確診1例X-SCID患者,進一步提高對IL2RG相關X-SCID認識,豐富了X-SCID相關的數據資料,為X-SCID的發現、診斷、治療及此類患兒家庭遺傳咨詢及產前診斷提供了依據。
