一測多評法同時測定山苦荬中3 種成分
2021-03-25 11:16:24黃健軍甘洋縈陳潔銀劉真亦
中成藥 2021年3期
黃健軍,梁 爽*,甘洋縈,陳潔銀,劉真亦
(1.廣西中醫藥大學,廣西 南寧 530001;2.玉林市食品藥品檢驗檢測中心,廣西 玉林 537000)
山苦荬Ixeridium chinense(Thunb.) Tzvel.為菊科小苦荬屬多年生草本植物,又名中華苦荬菜、山苦菜、小苦菜等,主要分布在北部、南部及東部省區,全草入藥,既可藥用又可食用,具有清熱解毒、消炎涼血、止痛消腫、抗腫瘤等功效,用于治療無名腫痛、腹腔膿腫、痢疾、闌尾炎、肺炎、關節炎等癥[1-2]。現代藥理學研究表明古羊藤有多種潛在的抗腫瘤、抗氧化、抗煙堿、抗病毒、降血脂、降血糖等活性[3-4]。山苦荬成分主要包含倍半萜類、三萜類、甾醇類、黃酮類等[4-7]。山苦荬作為廣西民間傳統瑤藥,其野生資源非常豐富,有報道對山苦荬中木犀草苷、綠原酸的含量進行測定[8-9]。經查閱文獻,當前國內外尚未見報道一測多評法同時測定山苦荬中綠原酸、咖啡酸和木犀草苷的含量。一測多評法基于多指標質量控制的研究思路,近年來獲得了業內專家學者的共識,只使用一個對照品實現對多種成分的同步測定,既降低了檢測成本,同時更簡便、全面地對中藥進行質量控制,提高了同步測定多種成分的工作效率[10-12]。基于上述因素,本研究擬對山苦荬的質量標準進一步完善,通過一測多評建立專屬性強的含量測定方法,建立與目前檢測技術相匹配的質量標準,以期為有效評價和控制山苦荬藥材質量及該藥材資源的開發提供參考。
1 材料
Agilent 1260 高效液相色譜儀(美國Agilent 公司);U3000 高效液相色譜儀(美國Thermo Fisher公司);XS-205Du 型電子分析天平(瑞士Mettler Toledo 公司);KQ-500GDV 超聲波清洗器(昆山市超聲儀器有限公司);501 超級恒溫水浴鍋(金壇市醫療儀器廠);Synergy 超純水(美國Millipore 公司。山苦荬藥材采自廣西各地區,藥材來源主要是采購和野外采集,具體見表1,經廣西中醫藥大學韋松基教授鑒定為菊科小苦荬屬植物中華小苦荬Ixeridium chinense(Thunb.) Tzvel.的全草。綠原酸(110753-201415,純度96.2%)、咖啡酸(110885-200102,純度100%)、木樨草苷(111720-201307,純度94.0%) 均購自中國食品藥品檢定研究院。乙腈、甲醇(色譜純,美國Fisher 公司);磷酸(優級純,批號20150327,天津市光復精細化工研究所);高效液相色譜議用水為超純水,其他所用試劑均為分析純。
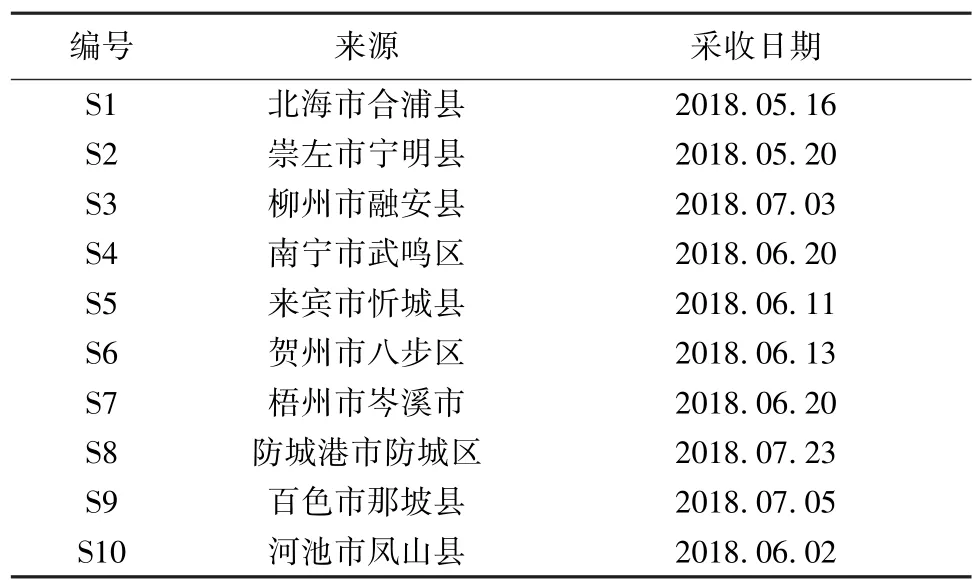
表1 樣品信息Tab.1 Information of samples
2 方法與結果
2.1 色譜條件 TYPE MGⅡSIZE-C18色譜柱(4.6 mm×250 mm,5 μm);流動相甲醇-0.2%磷酸,梯度洗脫,程序見表2;體積流量1.0 mL/min),梯度洗脫;柱溫25 ℃;檢測波長330 nm。

表2 梯度洗脫程序Tab.2 Gradient elution programs
2.2 溶液制備
2.2.1 混合對照品溶液 精密稱取對照品綠原酸11.59 mg、咖啡酸6.72 mg、木樨草苷13.60 mg,分別置于不同的25 mL 量瓶中,加適量甲醇超聲(500 W,40 kHz) 溶解并稀釋至刻度,搖勻,再分別精密量取上述相應對照品溶液1.5、1.5、6.0 mL,置同一10 mL 量瓶中并用甲醇定容,搖勻,即得。
2.2.2 供試品溶液 取山苦荬樣品約2 g,精密稱定,置于100 mL 錐形瓶中,精密加入70% 甲醇30 mL,稱定質量,超聲(500 W,40 kHz) 提取60 min,放冷,用70% 甲醇補足減失的質量,濾過,取續濾液過0.45 μm 微孔濾膜,即得。
2.3 方法學考察
2.3.1 系統適應性與專屬性試驗 分別精密吸取混合對照品、供試品溶液各5 μL,在“2.1” 項色譜條件下進樣,供試品溶液中綠原酸、咖啡酸和木犀草苷色譜峰與對照品色譜峰保留時間一致,并與其他共存成分的分離度大于1.5,理論塔板數不低于5 000。色譜圖見圖1。
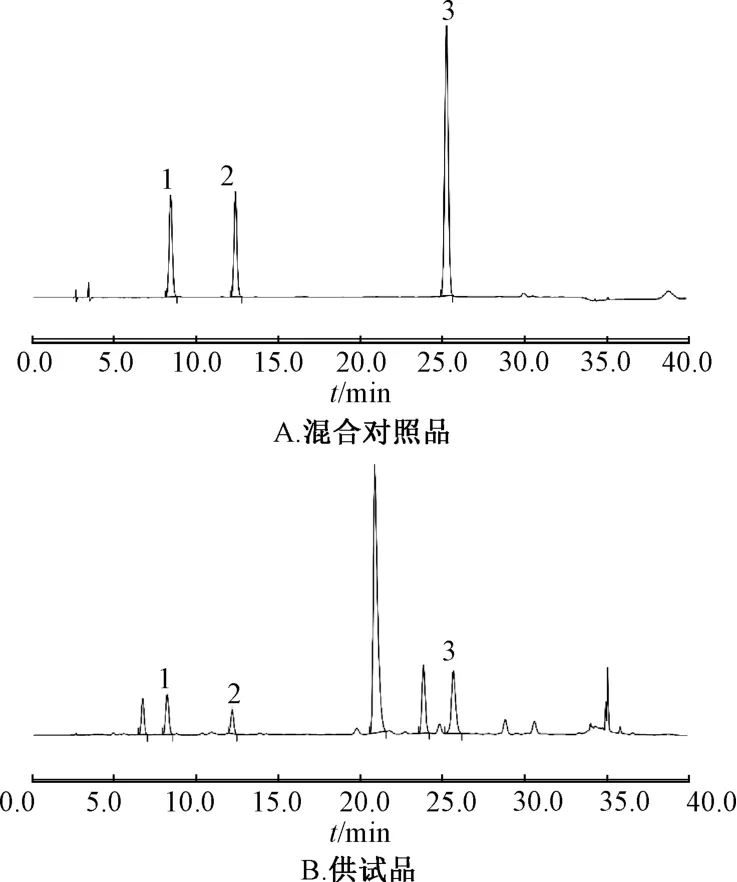
圖1 各成分HPLC 色譜圖Fig.1 HPLC chromatograms of various constituents
2.3.2 線性關系考察 分別精密吸取“2.2.1”項下混合對照品溶液0.4、0.6、0.8、1.0、1.2、1.6 mL,分別置2.0 mL 量瓶中,加70%甲醇定容至刻度,搖勻,制成系列混合對照品溶液,在“2.1” 項色譜條件下進樣,以目標峰峰面積為縱坐標(Y),質量濃度為橫坐標(X),進行回歸,得綠原酸、咖啡酸、木樨草苷回歸方程分別為Y=0.288X+0.057 (r=0.996 8)、Y=0.467X+0.034(r=0.997 1)、Y=0.174X+1.571 (r=0.991 2),各成分在各自范圍內線性關系良好。
2.3.3 精密度試驗 取山苦荬樣品(S4 號) 約2 g,按“2.2” 項下方法制備供試品溶液,在“2.1” 項色譜條件下連續進樣6 次,測得綠原酸峰面積平均值為15.62,RSD 為2.0%;咖啡酸峰面積平均值為5.47,RSD 為2.1%;木樨草苷峰面積平均值為31.04,RSD 為2.1%,表明儀器精密度良好。
2.3.4 重復性試驗 取S4 號山苦荬樣品6 份,每份約2 g,按“2.2” 項下方法制備供試品溶液,在“2.1” 項色譜條件下進樣,測得綠原酸含量平均值為1.17 mg/g,RSD 為1.7%;咖啡酸含量平均值為0.21 mg/g,RSD 為2.3%;木樨草苷含量平均值為2.51 mg/g,RSD 為2.3%,表明該方法重復性良好。
2.3.5 穩定性試驗 取“2.3.4” 項下制備的供試品溶液,在“2.1” 項色譜條件下,分別于0、2、4、8、12、24 h 進樣,測得綠原酸含量平均值為1.15 mg/g,RSD 為5.1%;咖啡酸含量平均值為0.21 mg/g,RSD 為4.8%;木樨草苷含量平均值為2.41 mg/g,RSD 為5.1%,表明供試品溶液在24 h 內穩定性良好。
2.3.6 加樣回收率試驗 取已測定含量的山苦荬樣品6 份,每份約1 g,精密稱定,另精密稱取對照品綠原酸6.86 mg、咖啡酸1.34 mg、木樨草苷16.24 mg,置200 mL 量瓶中,加適量70%甲醇超聲使溶解,并稀釋至刻度,搖勻,即得混合對照品貯備液。分別精密加入混合對照品貯備液30 mL,按“2.2” 項下方法制備供試品溶液,在“2.1”項色譜條件下進行測定,記錄山苦荬加樣回收樣品溶液中綠原酸、咖啡酸、木樨草苷的峰面積并計算回收率,結果見表3。
2.4 相對校正因子計算 精密吸取“2.2.1” 項下混合對照品溶液,依法測定咖啡酸、木樨草苷的峰面積,以綠原酸為內參物,按照相對校正因子計算公式[fsi=fs/fi=(As/Cs)/ (Ai/Ci)] (式中As為內參物綠原酸對照品峰面積,Cs為內參物綠原酸對照品濃度,Ai為待測成分咖啡酸或木樨草苷對照品峰面積;Ci為待測成分咖啡酸或木樨草苷對照品濃度。) 計算常咖啡酸、木樨草苷的相對校正因子,結果見表4。
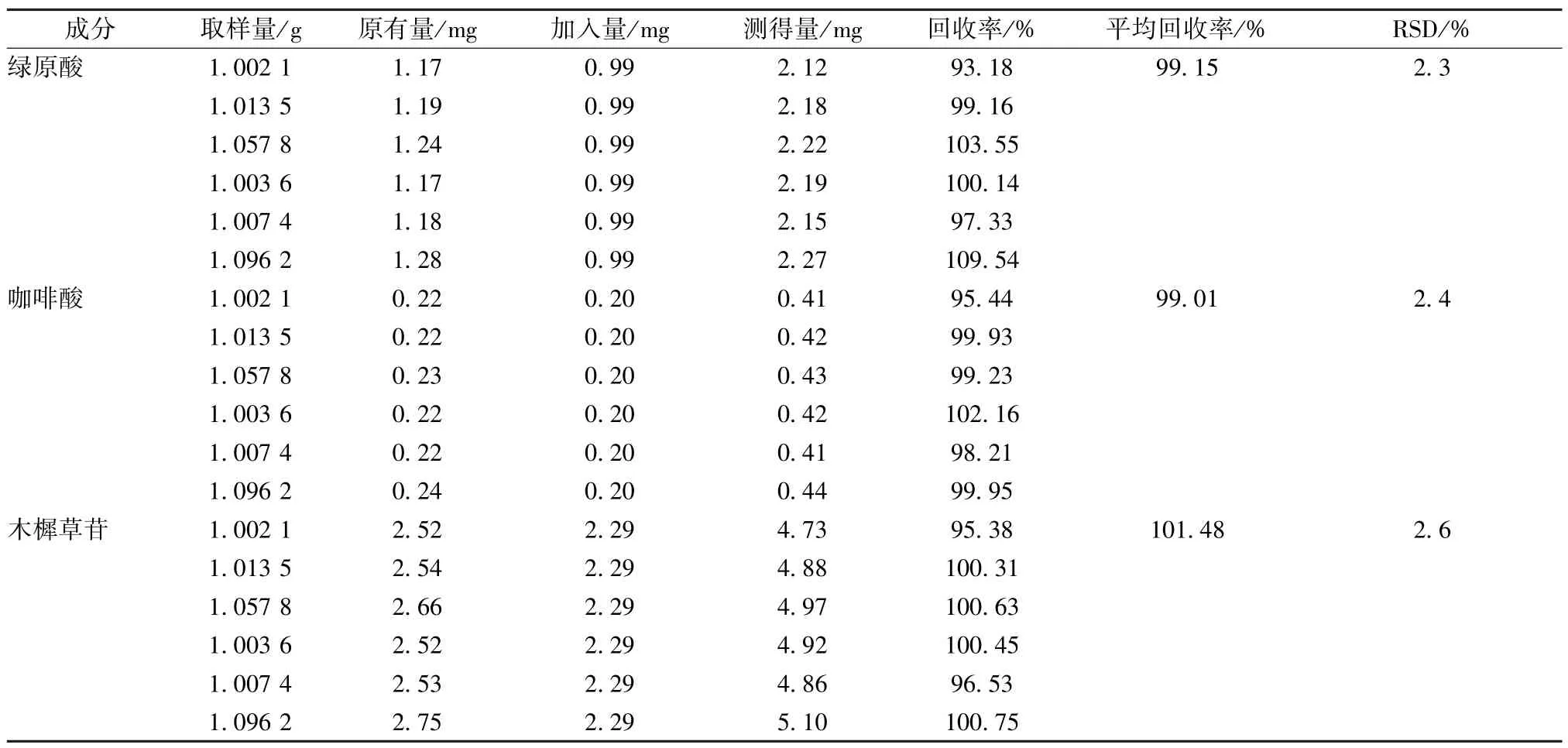
表3 各成分加樣回收率試驗結果(n=6)Tab.3 Results of recovery tests for various constituents (n=6)
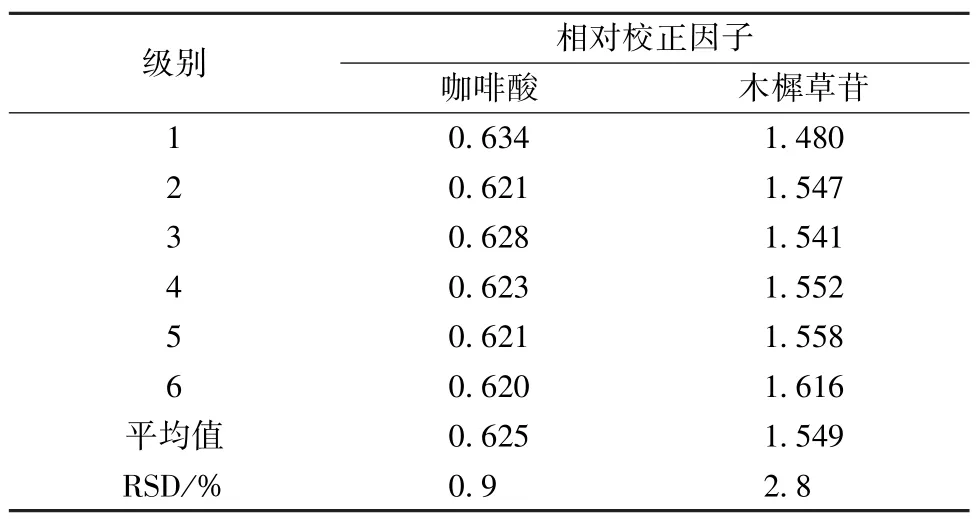
表4 各成分相對校正因子Tab.4 Relative correction factors of various constituents
2.5 系統耐用性考察
2.5.1 進樣量 精密吸取“2.2.1” 項下混合對照品溶液,在“2.1” 項色譜條件下進樣,考察4、5、6 μL 不同進樣量對相對校正因子的影響。結果顯示,不同體積流量對相對校正因子影響程度不明顯,咖啡酸和木犀草苷相對校正因子值的RSD 分別為0、0.2%,相對保留時間RSD 均為0。表明該方法耐用性良好,結果見表5。
2.5.2 儀器、色譜柱 選擇ThermoU3000 高效液相色譜儀、Agilent 1260 高效液相色譜儀,以及YPE MGⅡSIZE -C18(4.6 mm×250 mm,5 μm)、Agilent Eclipse plus-C18(4.6 mm × 250 mm,5 μm)、WatersAtlantis T3色譜柱(4.6 mm×250 mm,5 μm)考察不同儀器、色譜柱對相對校正因子的影響。結果顯示,不同儀器、色譜柱對相對校正因子影響程度在可接受范圍內,咖啡酸和木犀草苷相對校正因子RSD 分別為2.4%、1.1%,結果見表6。
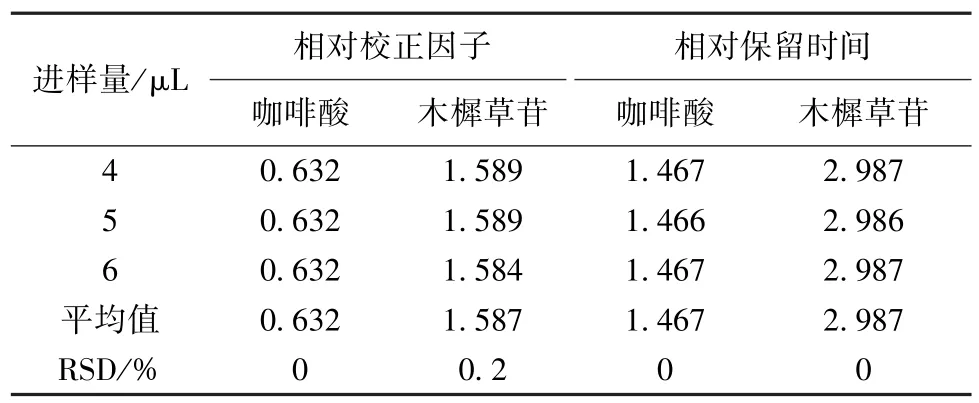
表5 不同進樣量的相對校正因子Tab.5 Relative correction factors or different injection volumes
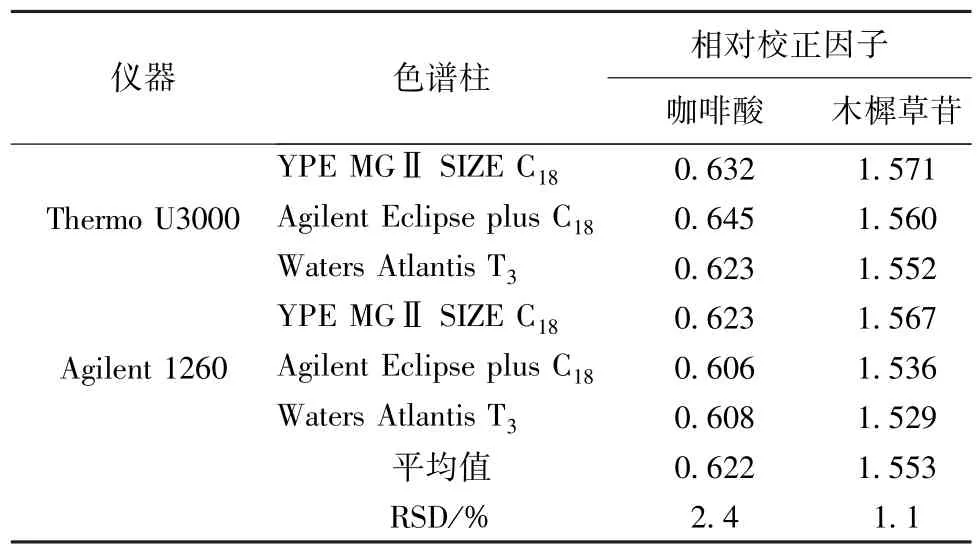
表6 不同儀器、色譜柱對相對校正因子的影響Tab.6 Effects of different instruments and chromatographic columns on relative correction factors
2.5.3 體積流量 精密吸取“2.2.1” 項下混合對照品溶液5 μL,在“2.1” 項色譜條件下進樣測定,考察0.9、1.0、1.1 mL/min 體積流量對相對校正因子的影響。結果顯示,不同體積流量對相對校正因子影響程度不明顯,咖啡酸和木犀草苷相對校正因子RSD 分別為0.3%、0.12%,相對保留時間RSD 分別為1.2%、5.1%,結果見表7。

表7 不同體積流量對相對校正因子的影響Tab.7 Effects of different volume flow rate on relative correction factors
2.5.4 柱溫 精密吸取“2.2.1” 項下對照品溶液5 μL,在“2.1” 項色譜條件下進樣,考察20、25、30 ℃不同柱溫對相對校正因子的影響。結果顯示,不同柱溫對相對校正因子影響程度不明顯,咖啡酸和木犀草苷相對校正因子RSD 分別為0.3%、0.5%,相對保留時間RSD 分別為1.0%、4.2%,結果見表8。

表8 不同柱溫對相對校正因子的影響Tab.8 Effects of different column temperature on relative correction factors
2.5.5 檢測波長 精密吸取“2.2.1” 項下對照品溶液5 μL,在“2.1” 項色譜條件下進樣,考察220、254、330 nm 檢測波長對相對校正因子的影響。結果顯示,不同檢測波長對相對校正因子影響程度不明顯,咖啡酸和木犀草苷相對校正因子RSD 分別為1.3%、5.7%,相對保留時間RSD 均為0,結果見表9。
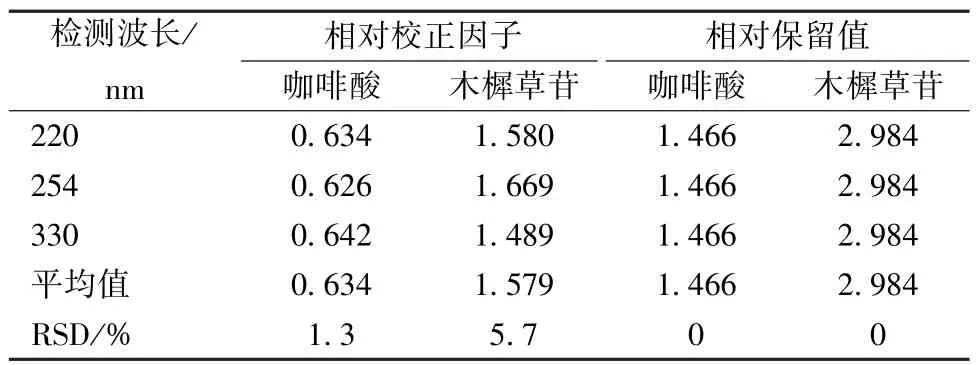
表9 不同檢測波長對相對校正因子的影響Tab.9 Effects of different detection wavelength on relative correction factors
2.6 待測色譜峰定位 分別考察相對保留值和保留時間差在不同儀器、色譜柱中的重復性,結果見表10。由表可知,咖啡酸和木犀草苷相對校正因子值波動較小,RSD 分別為1.4%、6.8%,故選擇綠原酸作為色譜峰定位指標。
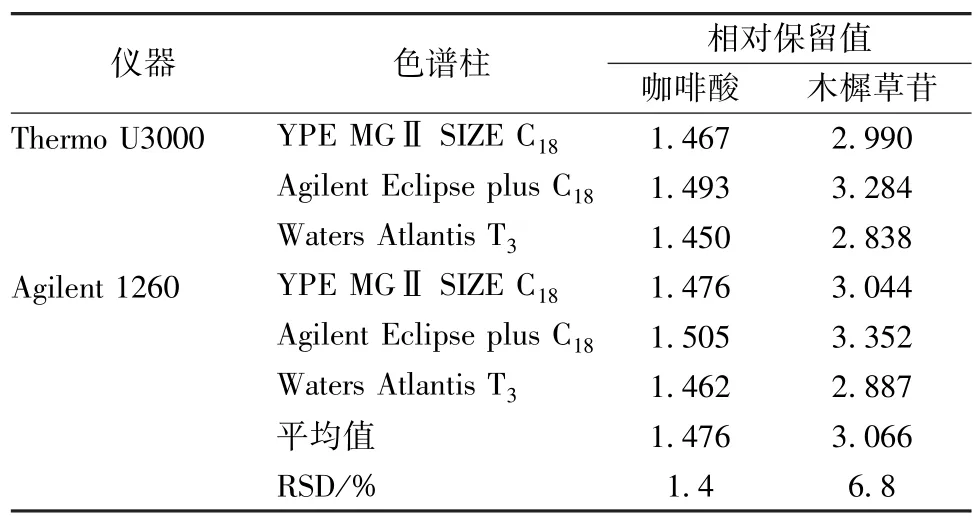
表10 不同儀器、色譜柱相對保留值的影響Tab.10 Effects of different instruments and chromatographic columns on relative retention values
2.7 一測多評法與外標法比較 經方法學優化和考察后,對10 批樣品提取物中的咖啡酸、木犀草苷的含量進行測定,為了考察一測多評方法的準確性,把一測多評法的測定結果與外標法測定結果進行比較,結果見表11。由此顯示,綠原酸含量較高的產地為S10、S6、S4,咖啡酸含量較高的產地為S10、S5、S6、S4、S2,木樨草苷含量較高的產地為S10、S5、S4、S6,表明S10、S6、S4 的山苦荬綠原酸、咖啡酸和木犀草苷的含量較高。
3 討論
參考文獻[13-18],本實驗分別選用0.1%磷酸-乙 腈、0.1% 磷 酸-甲 醇、0.2% 磷 酸-乙 腈、0.2%磷酸-甲醇、水-甲醇等;用0.1%磷酸時由于出峰時間遲,分離效果差,故排除0.1%磷酸;在選用乙腈和甲醇時,由于乙腈在分離時鋒形較窄且分離效果較差,出來的峰也較少;由于水的極性較大,所以考慮加酸,選擇0.2%磷酸-甲醇采用梯度洗脫時分離效果較好。分別考察了不同濃度甲醇、不同料液比、不同提取方法、不同提取時間,發現用70%甲醇超聲提取后所得到的峰基線平穩,色譜圖中檢測成分較多且峰形較好。用70% 甲醇不同體積進行測定,發現30 mL 所提取出來的含量較高。進一步用70%甲醇30 mL 分別用回流和超聲提取,發現兩者結果誤差不大,為了節省時間,故選擇回流。以70% 甲醇為提取溶劑提取30~120 min,結果發現60 min 時提取含量最高。本實驗對確定的對照品和藥材粉末以確定的色譜條件進行全波長掃描時發現在330 nm 均有最大吸收峰,保留時間一致,基線較平穩,色譜峰形較好,故選擇其作為檢測波長。
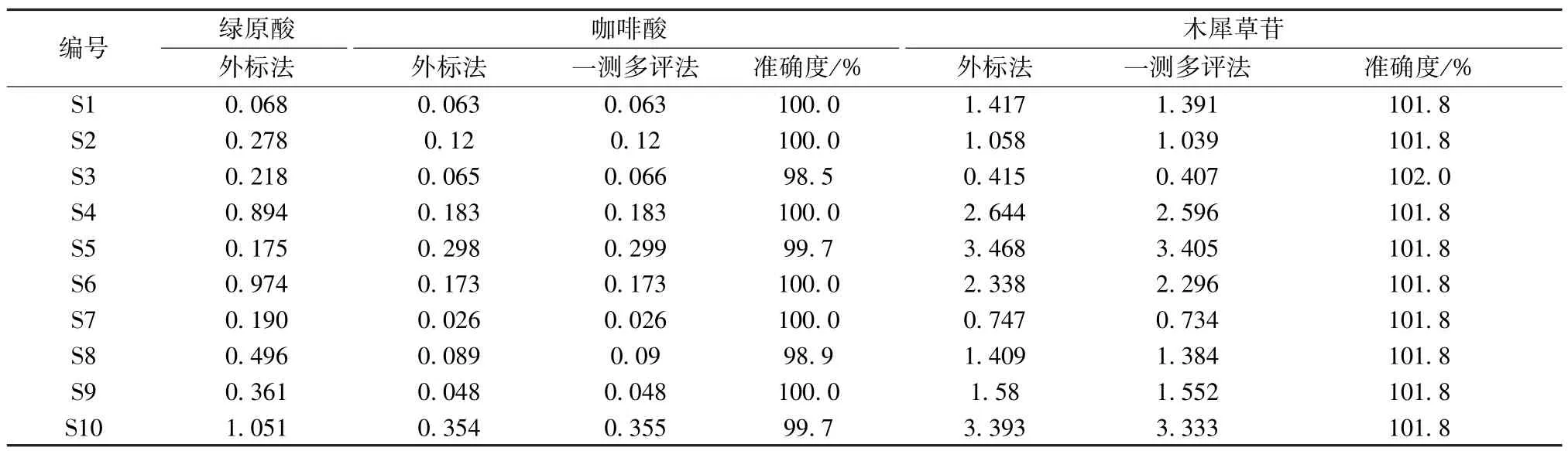
表11 一測多評法和外標法所得結果比較(mg/g)Tab.11 Comparison of results obtained by QAMS method and external standard method (mg/g)
山苦荬中由于成分較多,在現有對照品的條件下所測得3 個含量中綠原酸對照品較易購買,且實驗發現,以綠原酸為內標物,在不同儀器和色譜柱上的保留時間差波動較小,均在5%以內,由于咖啡酸含量較低,不宜進行系統性分析,而木樨草苷的保留時間大于5%,不宜進行待測組分色譜峰的定位。故選擇綠原酸作為內標。本實驗考察了不同儀器、色譜柱、體積流量、柱溫、檢測波長對各成分相對校正因子的重復性,用于驗證一測多評法在山苦荬質量控制中的可行性和技術適應性。結果表明,不同條件下相對校正因子的重復性均較理想。
針對在只使用單一對照品如何正確定位山苦荬中所測得其他2 種成分色譜峰的問題,選擇各成分之間的保留時間差、相對保留值、分離度作為定位標準,并在不同儀器、色譜柱下對二者進行考察。結果表明,相對保留值能更精準地定位色譜峰。本實驗建立一測多評法同時測定山苦荬綠原酸、咖啡酸和木樨草苷的含量。該方法簡單、快速、準確,相對校正因子具有良好的可信度和準確度。能在對照品短缺的情況下用于含量測定,同時可減少溶劑消耗和分析時間,更有利于環境保護。