洗滌用品及表面活性劑中活性物含量的測定
2021-03-30 08:17:00薛偉
中國洗滌用品工業 2021年2期
薛 偉
(1.中輕日用化學檢驗認證有限公司,山西太原,030001;2.全國表面活性劑和洗滌用品標準化技術委員會,山西太原,030001;3.國家洗滌用品質量監督檢驗中心,山西太原,030001)

洗滌用品是指通過洗凈過程用于清洗的專門配制的產品,其中起清潔作用的物質被稱之為“活性物”[1]。通常,“活性物”是指在洗滌產品中顯示規定活性的全部表面活性劑。根據產品類別的不同,有時候也被稱之為“有效物”,例如,皂類產品。為了保證洗滌用品的效能,對其進行質量檢測是十分有必要的。在諸多的檢測項目中,作為主要作用成分的活性物,其分析測試是不可或缺的。洗滌用品種類繁多,不同的洗滌用品,其活性物組成、含量等各不相同,檢測方法也各有不同。洗滌用品中活性物含量的測定方法包括乙醇溶解法[2]、差減法[3]、離子交換法[4]、電位滴定法[5-6]、兩相滴定法[7]等。作為洗滌用品活性物中的主要成分——表面活性劑,包含陰離子型、陽離子型、非離子型、兩性等多種,其活性物含量的測試方法根據表面活性劑類別的不同千差萬別。表面活性劑中活性物含量的測試方法包括兩相滴定法[8-9]、柱分離法[10]、電位滴定法[5-6]、差減法[11-12]、萃取法[12-14]、乙醇溶解法[13]、非水酸堿滴定法[15]等。
然而,無論是洗滌用品還是表面活性劑,現行標準體系中僅在相應的產品標準中指明了對應產品的活性物測試方法,而沒有一個較為系統的方法標準。鑒于活性物指標的重要性,以及現行標準中活性物測試方法相對較為分散,本文分別對洗滌用品和表面活性劑兩大類產品中活性物含量的測試方法進行系統介紹,以便于業內人員更加全面地了解活性物測試的各種方法,同樣也使分析測試技術人員的工作更加便捷、省時。
1 洗滌用品中活性物含量的測定
洗滌用品中活性物通常包括多種表面活性劑,因此,通常又將其稱之為“總活性物”。表1中列出了目前市場上常見的各種洗滌用品中總活性物含量的測定方法以及技術指標。
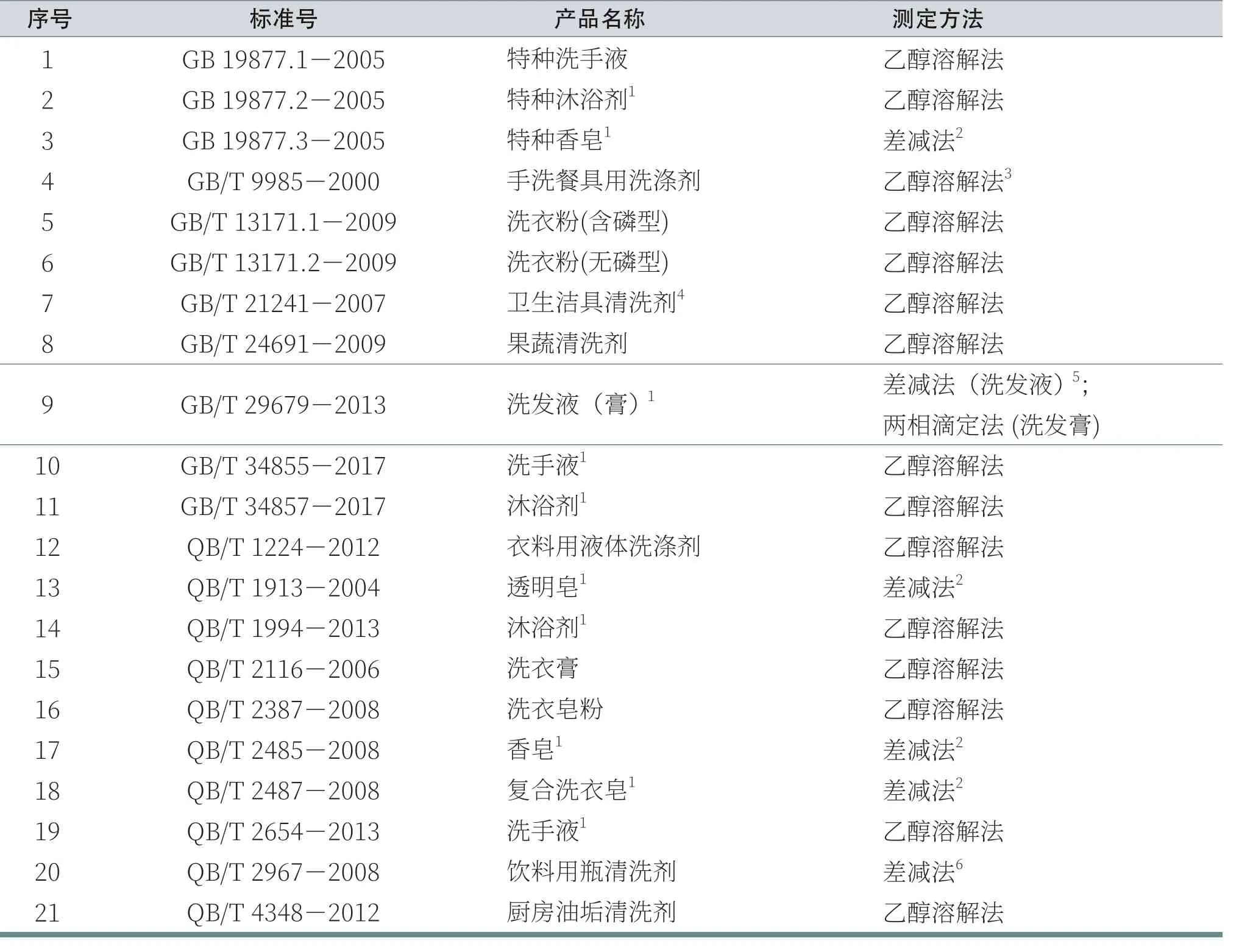
表1 不同洗滌用品中總活性物含量的測定方法及技術指標

續表1 不同洗滌用品中總活性物含量的測定方法及技術指標
1.1 乙醇溶解法
乙醇溶解法是目前洗滌用品中總活性物含量測定的最主要的方法,包括兩種:A法和B法。其中,A法的測定結果包含各種表面活性劑和水助溶劑,例如對甲苯磺酸鹽;B法則不包括水助溶劑,可以反映更加真實的活性物含量。由于B法使用毒性較大的氯仿,對操作人員及環境損害較大,且耗時較長,通常測試方法選擇A法,只有當客戶要求測試結果剔除水助溶劑時才會選擇使用B法。該方法適用于大多數洗滌用品中總活性物含量的測定。乙醇溶解法過程示意圖如圖1所示。
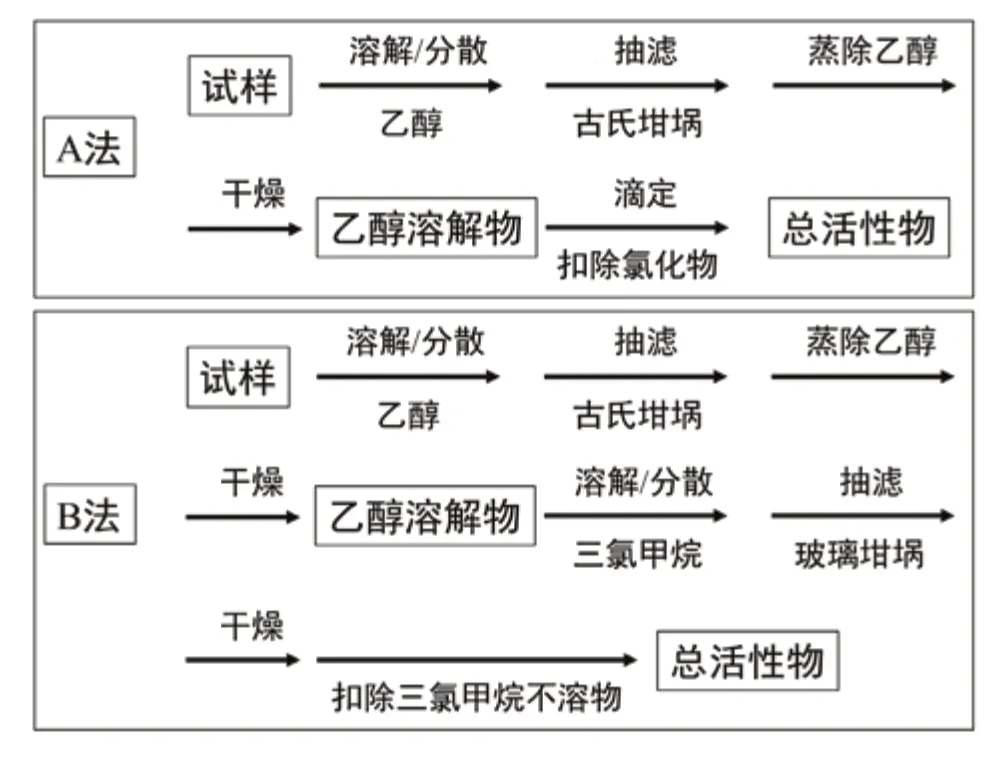
圖1 乙醇溶解法測定總活性物示意圖
原理 A法:用乙醇萃取樣品,定量乙醇溶解物含量,扣除其中的鹽分含量(以氯化鈉計,下同)后,即得到總活性物含量。鹽分作為洗滌用品中主要的無機助劑,其存在對產品的粘度等性能有影響,但是對清潔效果并無明顯的作用,不能提高洗滌用品的活性,因此需要扣除。B法:用乙醇萃取樣品,定量乙醇溶解物含量,然而用三氯甲烷進一步萃取該乙醇溶解物,扣除不溶物含量(包括水助溶劑等)即得總活性物含量。
注意事項 ①古氏坩堝的制備,需要依次沉積尺寸不同的酸洗石棉,方可得到可信度較高的測試結果。如果僅沉積顆粒度較大的酸洗石棉,可能會使試樣中尺寸較小的不溶物穿過,導致測試結果偏高;如果僅沉積顆粒度較小的酸洗石棉,會使抽濾過程的速度顯著減緩,時間大幅延長。此外,亦不能用玻璃坩堝代替古氏坩堝,因為在抽濾過程中容易發生穿濾或堵塞等現象。②A法中氯化鈉含量的測定,是以硝酸銀標準溶液通過化學滴定法測得的,指示劑選擇鉻酸鉀溶液。對于常規樣品,其滴定終點顏色由黃色變為橙色。但是,個別樣品是含有顏色的,例如衛生潔具清洗劑,在這種情況下,滴定過程中顏色發生突變時即可認定為滴定終點。
1.2 差減法
差減法是總活性物含量測定早期使用的方法,由于其操作步驟較多,耗時費力。在保證實驗結果相當的前提下,后來逐步精簡為乙醇溶解法。
目前,皂類產品[16-18]總有效物的測定通常是通過差減法測定的。皂類產品的總有效物是用乙醇萃取樣品,定量乙醇溶解物含量,扣除其中的游離苛性堿以及鹽分含量后計算可得。皂化過程中,存在有過量的游離苛性堿(以NaOH計),在計算總有效物時需將其扣除。其操作過程流程圖見圖2。

圖2 差減法測定總活性物流程示意圖
此外,洗發液和飲料用瓶清洗劑也是通過差減法進行測定的。洗發液產品的總有效物是先通過烘箱法得到固含量,扣除其中的乙醇不溶物和鹽分后,即可得到總有效物。飲料用瓶清洗劑的總活性物含量與洗發液類似,由固含量扣除乙醇不溶物、鹽分含量和酸/堿量后計算得到。飲料用瓶清洗劑總活性物測定流程見圖3。
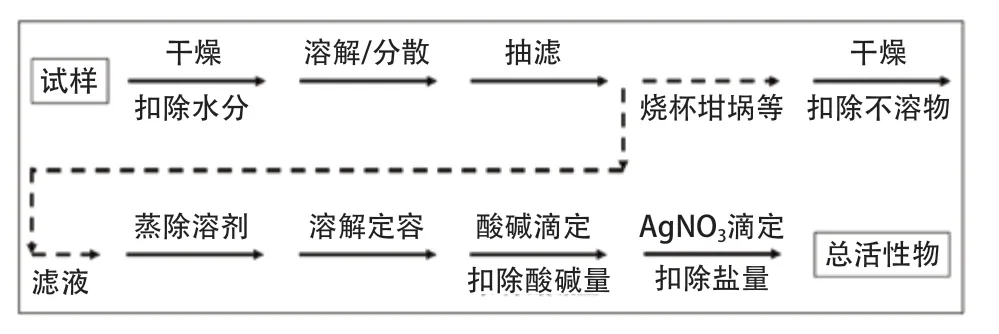
圖3 差減法測定飲料用瓶清洗劑中總活性物流程示意圖
1.3 離子交換法
離子交換法主要是用來測定洗滌用品中非離子表面活性劑的含量,例如乙氧基化烷基酚、聚乙氧基化脂肪醇、烷酰二乙醇胺等。
將樣品溶解于乙醇后,依次通過陽離子交換樹脂柱和陰離子交換樹脂柱,離子型表面活性劑在經過樹脂柱后與樹脂發生離子交換,保留在樹脂柱上,而非離子表面活性劑則不發生離子交換,會隨著溶劑流出。蒸除溶劑后,通過重量法即可得到其中非離子表面活性劑的含量。
注意事項 ①離子交換樹脂前處理過程中,前期樹脂處理必須充分,洗滌必須完全,否則將會隨著樣品乙醇溶解液一起流出,使得非離子表面活性劑含量的測定結果偏高。②填充柱中應盡量避免氣泡的存在,氣泡的存在會導致離子交換過程不完全,使得測試結果的有效性下降。
1.4 兩相滴定法
洗發膏中總活性物含量的測定是通過兩相滴定法進行的。兩相滴定法適用于測定洗滌用品和離子型表面活性劑中活性物的含量,尤以在離子型表面活性劑中的應用更為廣泛,詳細介紹見2.1。
1.5 電位滴定法
作為一種相對新穎的方法,在現行洗滌用品的產品標準中尚未見電位滴定法測定洗滌用品中總活性物含量的報道。根據國家綠色化發展的方針政策,隨著各個行業環保要求的進一步提高,電位滴定法應運而生。作為兩相滴定法的替代方法,該方法不使用有機溶劑,也不需加入酸性混合指示劑,大幅度降低了污染物的排放量,具有顯著的優勢。具體原理等詳見2.2。
2 表面活性劑中活性物含量的測定
關于表面活性劑,離子型表面活性劑中活性成分通常被稱之為“活性物”,而非離子、兩性表面活性劑則更多被稱之為“主組分”,在這里我們將其統稱為“活性物”。表2列出了不同常見表面活性劑中活性物含量的測定方法及對應的技術指標。
部分表面活性劑產品標準中未將“(總)活性物”設置為檢測指標,未列入該表格,如GB/T17829《聚乙氧基化脂肪醇》、GB/T 19464《烷基糖苷》、QB/T 2118《兩性表面活性劑 十一烷基咪唑啉》等。
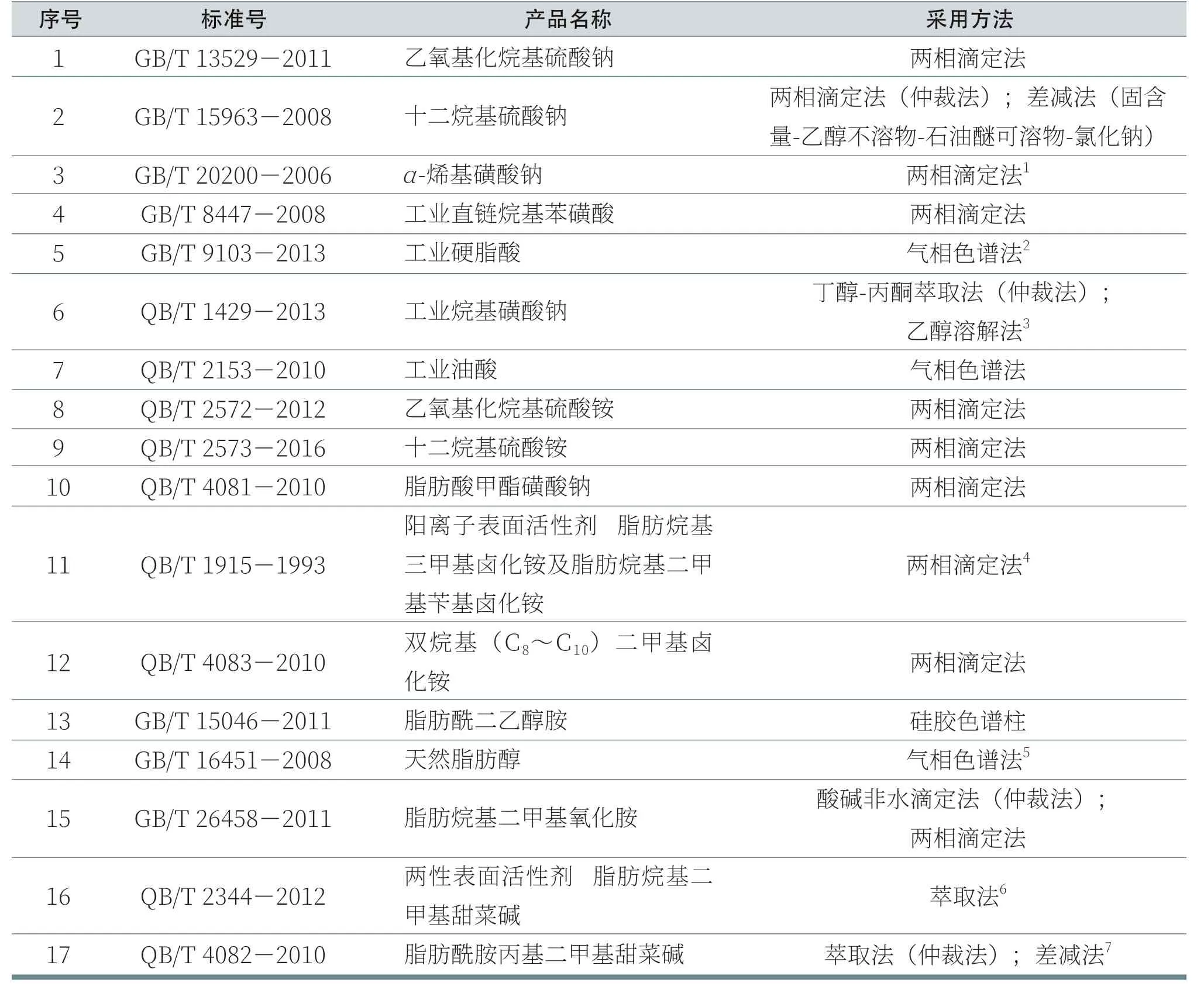
表2 不同表面活性劑中活性物含量的測定方法及技術指標
2.1 兩相滴定法
根據待測樣品中所含表面活性劑類型的不同,兩相滴定法又可以分為兩種:陰離子活性物含量的測定[8]和陽離子活性物含量的測定[9]。
原理 在水和有機溶劑(通常為三氯甲烷,亦可用二氯甲烷替代)的兩相介質中,并且酸性混合指示液存在下,用陽離子表面活性劑氯化芐蘇鎓(或陰離子表面活性劑十二烷基硫酸鈉)滴定,測定陰離子活性物(或陽離子活性物)。在測定陰離子活性物的反應過程中,陰離子活性物和陽離子染料生成鹽,此鹽溶解于三氯甲烷中,使三氯甲烷層呈粉紅色。滴定過程中,水溶液中所有陰離子活性物與氯化芐蘇鎓反應完,氯化芐蘇鎓取代陰離子活性物-陽離子染料鹽內的陽離子染料(溴化底米鎓),因溴化底米鎓轉入水層,三氯甲烷層紅色褪去。稍過量的氯化芐蘇鎓與陰離子染料(酸性藍-1)生成鹽,溶解于三氯甲烷層中,使其呈藍色。以氯化芐蘇鎓滴定乙氧基化烷基硫酸鈉為例,滴定過程中體系顏色變化如圖4所示。
從圖4可以看出,在滴定初始階段,乳化嚴重,分層非常困難(0號量筒)。隨著滴定過程的進行,分層變得較為容易,下層有機相的顏色由粉紅色逐漸變為淺灰-粉紅色(4號量筒),當粉紅色消失變為淺灰藍色時(5號量筒)為滴定終點。繼續滴定,有機相顏色則變為藍色(6號量筒)。但是,上層水相的顏色在整個過程中變化相對較小。
在測定陽離子活性物的反應過程中,在有陽離子染料和陰離子染料混合指示劑存在的兩相(水-氯仿)體系中,用陰離子表面活性劑標準溶液滴定樣品中的陽離子活性物。樣品中的陽離子表面活性劑最初與陰離子染料反應生成鹽而溶于三氯甲烷層,使其呈藍色。滴定中,陰離子表面活性劑取代陰離子染料,在終點時與陽離子染料生成鹽,使三氯甲烷層呈淺灰-粉紅色。以十二烷基硫酸鈉滴定十八烷基三甲基氯化銨為例,滴定過程中體系顏色變化如圖5所示。
從圖5可以看出,在滴定初始階段,分層非常困難(0號量筒)。隨著滴定過程的進行,分層變得較為容易,下層有機相的顏色由藍色逐漸變為淺藍色(4號量筒),當藍色褪去變為淺灰-粉紅色時(5號量筒)為滴定終點。繼續滴定,有機相顏色則變為粉紅色(6號量筒)。上層水相的顏色由最初的藍色變為了黃綠色。

圖4 氯化芐蘇鎓測定乙氧基化烷基硫酸鈉含量過程顏色變化

圖5 十二烷基硫酸鈉測定十八烷基三甲基氯化銨含量過程顏色變化
測定陽離子表面活性劑(如脂肪烷基三甲基鹵化銨)所使用的滴定劑還可以選擇四苯硼鈉溶液[19],此時指示劑則需要調整為溴酚藍,指示劑的添加量大幅減少至0.4 mL。該過程需要在強堿性環境下進行,當有機相中藍色消失時即為滴定終點。以四苯硼鈉滴定苯扎氯銨為例,滴定過程中體系顏色變化如圖6所示。
從圖6中可以看出,在滴定初始階段,分層較為容易,上層水相和下層有機相均為藍色(0號量筒)。隨著滴定過程的進行,分層變得更加容易,下層有機相的顏色由藍色逐漸變為深藍色(如3號量筒),后藍色逐漸變淺(6號量筒),最終褪去變為無色時(7號量筒)為滴定終點。繼續滴定,有機相顏色保持無色不再發生變化(8號量筒)。在該過程中,上層水相的顏色發生了顯著變化,由最初的藍色變淺為接近無色(4號量筒,此時并未達到滴定終點),繼續滴定水相顏色逐漸加深至紫色(6~8號量筒)。
注意事項 ①對于部分表面活性劑,按照上述方法進行活性物含量的測定時,可能滴定終點會難以識別,水與有機溶劑兩相分層速度不理想的情況,例如α-烯基磺酸鈉,此時需要對滴定過程中各試劑添加量進行調整,以期取得更加理想的測試結果。②滴定過程中,振搖應充分,使反應過程更加充分,以便測試結果可以更加真實地反映樣品情況。③對于一些分子量較大的陽離子表面活性劑,其在水中的溶解性能較差,在測試過程中需使用充分溶解的試樣溶液,以保證測試結果的真實有效性。必要時,可適度增加異丙醇用量來改善其溶解狀況。④當使用四苯硼鈉作為滴定劑時,四苯硼鈉標準溶液的標定可以通過已知純度的氯化芐蘇鎓測定,但是不能通過經由十二烷基硫酸鈉標定的氯化芐蘇鎓標準溶液。這是因為氯化芐蘇鎓與十二烷基硫酸鈉反應生成沉淀時的化學計量比不是嚴格的1:1(1:1.03~1.04),二次標定后量值傳遞可能導致偏高的測定結果。
2.2 電位滴定法
該方法為兩相滴定法的替代方法,主要適用于離子型表面活性劑或按配方制造的產品中活性物含量的測定。

圖6 四苯硼鈉測定苯扎氯銨含量過程顏色變化
原理 表面活性劑離子選擇電極對離子型表面活性劑有選擇性響應,在滴定過程中,電極電位將隨著表面活性劑濃度的改變而變化。在滴定終點處,體系中游離的表面活性劑類型發生顯著變化,電極電位發生突躍[20-23]。在測定含有陰離子表面活性劑的樣品時,以陽離子表面活性劑標準溶液為滴定劑(常選擇氯化芐蘇鎓),與體系中陰離子表面活性劑發生沉淀反應,逐步降低陰離子表面活性劑在溶液中濃度。當體系中陰離子表面活性劑被標準滴定溶液完全耗盡,體系中電極電位將會呈現出明顯的突躍(對滴定標準溶液消耗體積-電極電位曲線進行求導即可判定),此時即為滴定終點。根據此時消耗的滴定劑用量,計算得到體系中陰離子活性物含量。在測定含有陽離子表面活性劑的樣品時,以陰離子表面活性劑標準溶液為滴定劑(常選擇十二烷基硫酸鈉),與體系中陽離子表面活性劑發生反應生成沉淀,使游離的陽離子表面活性劑濃度逐步降低。當滴定體系中陽離子表面活性劑與滴定劑反應完成后,離子選擇電極指示的電極電位將會發生顯著變化,在滴定曲線上的突變點即為滴定終點。根據此時消耗的滴定劑用量,計算得到陽離子活性物含量。典型的陰離子滴定劑通過電位滴定法測定陽離子表面活性劑含量的示意圖[22]見圖7。

圖7 電位滴定法測定陽離子表面活性劑含量示意圖
從圖7可以看出,隨著滴定的進行,電位值先緩慢下降,然后在滴定終點附近顯著下降,最終趨向于恒定不變,曲線形狀為“S”型曲線的鏡像。由陽離子滴定劑測定陰離子表面活性劑含量時,曲線變化趨勢剛好相反,呈現為“S”型曲線。滴定曲線中存在有明顯的電位突躍,一階導數曲線的突出峰值處即對應于滴定終點體積。
該方法的測定結果與兩相滴定法相當。與兩相滴定法相類似,尿素、乙二胺四乙酸鹽、羧甲基纖維素、氯化鈉、硫酸鈉等洗滌用品中常見的添加成分對實驗結果的真實有效性無干擾。與兩相滴定法相比,該方法是在水溶液中進行的,不使用氯仿等有機溶劑,也不使用酸性混合指示劑,可顯著降低環境負荷,同時降低了實驗成本,符合國家綠色化發展的政策。此外,該滴定過程中所使用的儀器為實驗室常見的酸度計,離子選擇電極可多次循環重復使用,具備了大幅推廣應用的基本條件。中國日化院研制的表面活性劑離子選擇電極可以同時響應陰離子表面活性劑和陽離子表面活性劑,已經達到了國際先進水平。
注意事項 ①離子選擇電極的使用及貯存需按照相關說明進行,否則,其壽命將會大幅縮短。②對于不同的表面活性劑,適宜測定的pH范圍不同,在輕工行業標準QB/T 4969和QB/T 4970中均已列出,在滴定時需要參考該pH范圍以便得到可靠的結果。
2.3 氣相色譜法
表面活性劑或用于制備表面活性劑的主要原材料,如工業硬脂酸[24]、天然脂肪醇[25]、脂肪烷基二甲基叔胺[26]、工業油酸[27]等,其活性物含量可通過氣相色譜法測定。根據待測試樣的性質,選擇合適的氣相色譜儀、色譜柱、色譜條件、前處理方式以及計算方法,即可得到其中活性物含量。色譜儀、色譜柱以及色譜條件的選擇以各色譜峰分離完全、不能重疊為依據。在同一測試條件下,一般是通過保留時間進行定性分析,定量分析則可以使用峰面積歸一化法、內標法、外標法等。
2.4 萃取法
甜菜堿類表面活性劑和工業烷基磺酸鈉中活性物含量的測定通常選用萃取法提取活性物,結合滴定法、重量法等其他方法計算得到活性物含量。通過萃取法提取脂肪烷基二甲基甜菜堿和脂肪酰胺丙基二甲基甜菜堿中活性物之后,結合電位滴定法測得其活性物含量。在工業烷基磺酸鈉萃取出活性組分之后,結合重量法,最后扣除其中鹽分含量即可得到活性物含量。不同的表面活性劑,所選擇使用的溶劑略有不同。對于脂肪烷基二甲基甜菜堿和脂肪酰胺丙基二甲基甜菜堿,所使用的萃取溶劑分別為正丁醇和正丁醇-乙醚混合溶劑,而工業烷基磺酸鈉所使用的溶劑為丁醇-丙酮混合溶劑。除了萃取法(仲裁法),工業烷基磺酸鈉中活性物含量還可以通過乙醇溶解法測得。脂肪酰胺丙基二甲基甜菜堿中活性物同樣可以通過差減法測得,即固含量扣除鹽分和游離胺。
2.5 其他方法
脂肪酰二乙醇胺是通過硅膠色譜法[10]測定的。其原理為:樣品經過硅膠色譜柱時,各種物質與填充硅膠的吸附能力強弱不同,然后用不同極性的溶劑洗脫,分別收集,定量分析即可得到活性物含量。通常,用石油醚-乙醚的混合溶劑沖洗脂肪酰二乙醇胺中殘留的甲酯,用丙酮或三氯甲烷-乙醇的混合溶劑沖洗脂肪酰二乙醇胺活性物。
脂肪烷基二甲基氧化胺中活性物含量的測定可以通過酸堿非水滴定法(仲裁法)和兩相滴定法進行[15]。鹽酸的醇標準溶液在非水條件下滴定脂肪烷基二甲基氧化胺,氧化胺中未反應的胺與碘甲烷發生季銨化反應,生成的季銨鹽在該條件下不能與鹽酸發生反應,因此可確定樣品中氧化胺的含量。注意事項:①季銨化過程需完全,否則測試結果偏高。②滴定需要在非水條件下進行,否則季銨鹽同樣可能參與反應過程,致使不可靠結果的出現。
通過差減法可以測定十二烷基硫酸鈉[11]和脂肪酰胺丙基二甲基甜菜堿[12]中活性物含量,需要說明的是,該方法不是這兩種物質中活性物含量測定的仲裁法。在測定十二烷基硫酸鈉中活性物含量時,需要先測定固含量,然后扣除石油醚可溶物、乙醇不溶物以及乙醇溶解物中的鹽分,即可得到其活性物含量。
3 展望
隨著社會的發展進步,新型表面活性劑陸續推向市場,盡管可以借鑒已有表面活性劑的檢測方法,但是嘗試進一步挖掘新的活性物含量的測試方法是很有必要的。同時,洗滌用品種類日益繁多,其配方也在發生著日新月異的變化,添加成分更加多元化,功能更加綜合化,消費定位更加層次化。在這種新形勢下,以現有方法體系為基礎,或進行適度修正,或尋找更加合適的新方法來測定其活性物含量,規避新型添加劑的干擾,依舊任重道遠。
