海藻糖合成酶結構及其抑制劑的研究進展
2021-04-06 08:10:28蔣志洋王金娥張婧瑜鄧鳴飛黃家興段紅霞
農藥學學報 2021年2期
關鍵詞:結構
蔣志洋, 韓 清, 王金娥, 朱 凱, 張婧瑜, 鄧鳴飛, 黃家興, 段紅霞
(中國農業大學 理學院 應用化學系 農藥創新中心,北京 100193)
海藻糖是由兩個分子葡萄糖通過α-1,1 糖苷鍵相連的非還原性二糖[1],其廣泛分布于昆蟲、細菌、真菌以及植物等眾多生物體內,發揮著重要的生物學功能。經研究表明,海藻糖不僅可作為儲能物質參與生物體的能量代謝[2-3],亦可作為結構物質參與昆蟲體壁和真菌細胞壁的形成過程[4-5],還可作為“毒力因子”決定病原菌的致病性[6-7],以及作為化學信號物質調控昆蟲的生長與發育[8-9]。
海藻糖合成酶是生物體內參與海藻糖合成的重要功能酶。在昆蟲體內,海藻糖-6-磷酸合成酶(trehalose-6-phosphate synthetase, TPS) 和海藻糖-6-磷酸磷酸酯酶 (trehalose-6-phosphate phosphatase,TPP) 是海藻糖合成酶的兩個獨立保守結構域;在細菌體內,分別對應為OtsA 和OtsB[10];在真菌酵母體內,海藻糖合成酶中的兩個催化亞基TPS1和TPS2 分別執行TPS 和TPP 的功能[2]。目前,在不同的生物體內已有5 條海藻糖合成途徑被闡明,分別為:海藻糖-6-磷酸合成酶和海藻糖-6-磷酸磷酸酯酶 (TPS-TPP/OtsA-OtsB/TPS1-TPS2) 途徑、麥芽低聚糖海藻糖合成酶-麥芽低聚糖海藻糖水解酶 (TreY-TreZ) 途徑、海藻糖合酶 (TreS) 途徑、海藻糖磷酸化酶 (TreP) 途徑及海藻糖糖基轉移酶 (TreT) 途徑[11-12]。其中海藻糖-6-磷酸磷酸合成酶和海藻糖-6-磷酸磷酸酯酶途徑是普遍被認可的途徑,該途徑中海藻糖-6-磷酸合成酶是其限速關鍵酶。其海藻糖-6-磷酸合成酶和海藻糖-6-磷酸磷酸酯酶途徑催化海藻糖生物合成的具體過程(圖1) 為:核苷酸葡萄糖 (NDPG) 和葡萄糖-6-磷酸(G6P) 在海藻糖-6-磷酸合成酶的催化下首先合成6-磷酸海藻糖 (T6P),隨后,由海藻糖-6-磷酸磷酸酯酶催化T6P 去磷酸化,最終生成海藻糖[13-15]。
有研究表明,海藻糖代謝通路在昆蟲和植物病原真菌的重要生理過程中存在著調控機制,如昆蟲海藻糖代謝可調控其幾丁質代謝通路。幾丁質是天然的線性糖胺聚糖[16],其作為昆蟲體壁的主要成分,在昆蟲生長發育過程中的舊表皮蛻去和新表皮形成中發揮著重要調控作用[17]。相關研究顯示,昆蟲海藻糖代謝路徑位于其幾丁質代謝通路上游,二者通過海藻糖酶相互偶聯 (圖2),即海藻糖在海藻糖酶催化下分解為葡萄糖,隨后,在己糖激酶催化下生成G6P,G6P 焦磷酸化生成UDP-N-乙酰氨基葡萄糖,隨即進入幾丁質生物合成通路,即UDP-N-乙酰氨基葡萄糖經幾丁質合成酶作用生成幾丁質,最終經幾丁質酶和β-N-乙酰己糖胺酶等作用,完成幾丁質降解,生成N-乙酰氨基葡萄糖[18]。海藻糖作為溝通這兩個通路的重要物質,其代謝過程可調控幾丁質的合成與代謝通路,從而影響昆蟲生長發育[19-20]。此外,海藻糖代謝還可調控植物病原真菌侵染過程。在對稻瘟病菌 Magnaporthe oryzae 侵染力調控中,存在著依賴于還原型煙酰胺腺嘌呤二核苷酸磷酸 (NADPH)的開關機制,該開關可被稻瘟病菌TPS1 調控。TPS1 缺失的稻瘟病菌其附著胞和侵染釘產生受阻,使其無法正常侵染[21-23]。有報道指出,真菌細胞壁的形成、菌絲的生長與其體內幾丁質酶存在著密切聯系,幾丁質酶異常可以導致植物病菌生長受阻、其致病能力下降[24-25]。因此,筆者推測在真菌中海藻糖酶也可能與幾丁質酶代謝相關,但尚未見相關文獻報道。
海藻糖代謝途徑憑借著其重要的生物學功能及其在哺乳動物體內缺失的優勢[26],在新型殺蟲劑、殺菌劑開發領域受到越來越廣泛關注。例如,以海藻糖酶為靶標的殺菌劑井岡霉素A (validamycin A) 已成功上市,且對水稻紋枯病、棉花猝倒病等具有良好的防效[27-28]。生物體內參與海藻糖合成的重要功能酶,如海藻糖-6-磷酸合成酶和海藻糖-6-磷酸磷酸酯酶也陸續被認為是新型殺蟲劑、殺菌劑研發的候選靶標[10,29],尤其是近年來有關海藻糖-6-磷酸合成酶和海藻糖-6-磷酸磷酸酯酶的結構生物學研究陸續取得較大進展,這為基于海藻糖相關酶靶標結構進行新農藥分子合理設計,進而開發綠色安全的新農藥品種奠定了重要生物學基礎。但迄今為止,尚未見有關海藻糖-6-磷酸合成酶和海藻糖-6-磷酸磷酸酯酶晶體結構及其抑制劑的相關綜述報道。鑒于此,本文主要綜述近年來關于海藻糖-6-磷酸合成酶和海藻糖-6-磷酸磷酸酯酶的晶體結構、底物結合模式及其酶抑制劑的研究進展,以期為以海藻糖合成酶為靶標的先導化合物的發現和新農藥活性分子合理設計提供指導和借鑒。
1 海藻糖合成酶晶體結構
1.1 海藻糖-6-磷酸合成酶晶體結構及其與底物結合模式分析
糖基轉移酶 (GTs) 是生物體內參與糖代謝的重要功能酶,負責多糖及其他糖類復合物的合成[30]。截至2020 年10 月,碳水化合物活性酶數據庫中(CAZy, http://www.cazy.org) 根據酶一級結構 (氨基酸序列) 保守性原則[31],將已知序列的GTs 劃分為111 個家族,即GT1~GT111。此外,根據GTs高級結構相似性,又將其劃分為GT-A、GT-B、GT-C 和GT-D 共4 個超家族[32-35]。通常大部分GTs 在形成三維結構時均采取GT-A 和GT-B 折疊型 (圖3)。這兩種折疊方式都可形成兩個典型的Rossmann 結構域 (包含β/α/β 型超二級結構),其差異主要體現在GT-A 折疊形成的兩個Rossmann結構域是彼此相鄰的,且聯系緊密;而GT-B 折疊形成的兩個相似的Rossmann 結構域往往更加獨立,聯系也更為靈活,且在兩結構域交界處常存在著縫隙[36-37]。
海藻糖-6-磷酸合成酶是GTs 中的重要成員。研究發現,TPS/OtsA 屬于GT20 家族,且采取GTB 折疊型,其催化活性位點存在于由兩個Rossmann結構域形成的縫隙中[36]。海藻糖-6-磷酸合成酶在海藻糖生物合成過程中負責將糖基供體底物NDPG的葡萄糖環部分轉移至糖基受體底物G6P 的葡萄己糖環上,并催化二者間α-1,1 糖苷鍵的形成。隨著蛋白質結晶學、X-射線晶體衍射以及核磁共振等技術日漸成熟,越來越多物種的海藻糖-6-磷酸合成酶晶體結構被解析。截至目前,真菌TPS1 和細菌OtsA 晶體結構均已有許多報道,但尚未見昆蟲TPS 晶體結構的報道。
1.1.1 細菌OtsA 晶體結構 目前,已有6 種細菌的17 個OtsA 蛋白晶體結構被解析,如表1 所示。其中大部分為人類病原細菌的OtsA 晶體,如大腸桿菌Escherichia coli 和耐熱分枝桿菌Mycobacterium thermoresistibile 的OtsA 晶體,分別簡稱為 EcOtsA和MtrOtsA 等,但尚未見植物病原細菌OtsA 晶體結構的報道。

表1 目前已報道的不同細菌OtsA 的晶體結構信息Table1 The reported crystal structures of different bacteria OtsAs
Gibson 等[14]于2002 年報道了首個海藻糖-6-磷酸合成酶三維晶體結構,即大腸桿菌OtsA 與小分子G6P、尿苷二磷酸 (UDP) 的復合物蛋白 (PDB號:1GZ5)。該研究指出:EcOtsA以四聚體形式存在,組成四聚體的單體結構之間是高度相似的,每個單體都包含有兩個Rossmann結構域,即N-端結構域和C-端結構域,在兩個結構域界面處有一明顯裂縫,OtsA 的催化中心就位于這一裂縫中。其N-端結構域核心是由7 條平行的β 鏈形成的三維結構,其核心兩側各有一條反向平行的β 鏈及6 個α 螺旋,而其C-端結構域則是由6 條β 鏈形成的核心和外側的7 個α 螺旋構成的。
Mendes 等[40]以EcOtsA 為模型,解析了耐熱分枝桿菌MtrOtsA 的四聚體結構 (PDB 號:5JIJ),其單體拓撲結構與EcOtsA 較相似,即其N-端結構域核心是由7 條平行的β 鏈及兩側反平行的β 鏈構成,該核心區域被外側8 個α 螺旋所包圍,而其C-端結構域則包含6 個平行β 鏈和9 個α 螺旋。位于C-端結構域最末端的α 螺旋可延伸至N-端結構域,并在這兩個結構域界面處產生紐結,這是GT-B 型折疊糖基轉移酶的普遍結構特征。然而,該研究也指出:雖然MtrOtsA 與EcOtsA 蛋白單體三維結構顯示出較高的相似性,但其相應序列比對結果顯示,與其他非分枝桿菌物種相比,MtrOtsA 蛋白四聚體界面處氨基酸殘基的保守性較低,而在分枝桿菌及其他近緣物種中,該位置殘基則呈現出高度保守性,這可能是由于不同物種海藻糖-6-磷酸合成酶在形成高級結構時,多肽鏈空間折疊方式存在著細微差異導致的。
1.1.2 真菌TPS1 晶體結構 與細菌OtsA 相比,被報道的真菌TPS1 晶體數量較少,截至目前,僅有3 種真菌的9 個TPS1 晶體被解析 (表2)。值得關注的是,Wang 等[41]于2019 年報道了首個農業病害微生物稻瘟病菌Magnaporthe oryzae MoTPS1的晶體結構,這為基于稻瘟病菌MoTPS1 結構進行新型農用殺菌劑合理化設計奠定了生物學基礎。
Miao 等[42]通過對已揭示三維結構的真菌TPS1 與細菌OtsA 多肽鏈的空間折疊方式進行比對發現:真菌TPS1 與細菌OtsA 高級結構總體相似度較高,即真菌TPS1 的C-端Rossmann 結構域內部也是由若干個平行β 鏈組成的β 折疊片構成的,其外側則被數個長短不一的α 螺旋包圍著,最末端α 螺旋也可跨越至N-端結構域繼續形成α 螺旋,并在兩結構域界面處產生紐結。如人類病原真菌白色念珠菌Candida albicans 的CaTPS1單體,C-端結構域就是由6 條β 鏈和8 個α 螺旋構成的,其α16 螺旋在氨基酸殘基Y457 處產生紐結,而稻瘟病菌MoTPS1 單體的C-端結構域則包含6 條β 鏈和10 個α 螺旋,其α18 螺旋于G252、T464 處產生紐結[41]。
當然,真菌TPS1 和細菌OtsA 三維結構也存在著顯著差異:如真菌TPS1 的N-端結構域表現為非典型Rossmann 結構域。有研究指出,CaTPS1單體N-端結構域中β1 鏈與一組伸出催化中心的反向平行β 折疊片 (β2-β3) 相連,而并非α 螺旋;同時該研究顯示,煙曲霉Aspergillus fumigatus AfTPS1B 單體也展現出同樣結構特點,而另一個AfTPS1A 單體中卻未觀察到該類似結構特征[42]。Wang 等[41]的研究同樣表明,MoTPS1 單體的N-端結構域中也存在著由β2 鏈和β3 鏈反向折疊形成的β-發夾結構,由于在細菌OtsA 中尚未觀察到類似發夾結構,因而推測該結構可能僅特異性地存在于真菌TPS1 中,并可作為區分真菌TPS1 和細菌OtsA 的顯著特征之一。
1.1.3 海藻糖-6-磷酸合成酶與糖基供體底物NDPG 的結合模式分析 在海藻糖-6-磷酸合成酶催化過程中,NDPG 發揮著糖基供體的作用,其可以與海藻糖-6-磷酸合成酶C-端結構域活性位點的氨基酸殘基結合 (圖4)。生物體內共存在5 種NDPG,分別為:腺苷二磷酸葡萄糖 (APDG)、尿苷二磷酸葡萄糖 (UPDG)、鳥苷二磷酸葡萄糖(GDPG)、胞苷二磷酸葡萄糖 (CDPG) 和胸苷二磷酸葡萄糖 (TDPG)。研究表明,大多數生物海藻糖-6-磷酸合成酶是以UDPG 為底物的,但少數物種酶對底物也存在著其他偏好,如MtrOtsA 和委內瑞拉鏈霉菌Streptomyces venezuelae SvOtsA 就是分別以ADPG 和GDPG 為最適供體底物的[39-40,43]。
分析已被解析的TPS1 蛋白結構,發現在3 種真菌TPS1——CaTPS1、AfTPS1 及MoTPS1 與底物NDPG 復合物晶體中,NDPG 葡萄糖環部分電子密度極低,以致于其結構不可見,這可能是由于TPS1 中NDPG 葡萄糖結合腔較大,柔性過高導致的[41-42]。除上述3 種真菌外,在其他細菌OtsA與NDPG 結合的二元復合物晶體中均可見葡萄糖環,且葡萄糖環與OtsA 結合的關鍵氨基酸殘基在不同生物體內是高度保守的,二者間分子互作模式也非常相似。如:大腸桿菌中,EcOtsA 以His154、Gln185、Asp361、Met363 和Asn364 等保守的氨基酸殘基與UDPG 的葡萄糖環通過氫鍵產生結合作用[14],在耐熱分枝桿菌MtrOtsA 中也可觀察到NDPG 的葡萄糖基與該酶活性中心的His168、Asp385、Gly386、Met387 及Asn388 等殘基形成氫鍵作用[40]。
NDPG 核糖部分及兩個磷酸根與OtsA/TPS1催化中心結合也是十分保守的,在不同物種海藻糖-6-磷酸合成酶晶體結構中均可以觀察到:NDPG 五元核糖環的羥基與該位點谷氨酸殘基以氫鍵產生相互作用;NDPG 兩個磷酸根結合于OtsA/TPS1 活性中心的正電結合腔中,該正電中心由一組保守的Lys-Arg 殘基對構成;此外,也可觀察到NDPG 磷酸根的羥基與該位點的亮氨酸殘基通過其主鏈氨基形成氫鍵相互作用。
然而,NDPG 堿基部分與海藻糖-6-磷酸合成酶的結合卻存在著一定物種間差異,如EcOtsA的Phe339 殘基可與UDPG 尿嘧啶的O4 和N3 分別形成氫鍵作用力[14],而在白色念珠菌和煙曲霉菌體內,UDPG 的尿嘧啶可分別與二者TPS1 中的Ile357 或Val364 殘基形成氫鍵[42]。Mendes 等[40]研究指出,MtrOtsA 在行使催化功能時,是不以UDPG 為糖基供體底物的,而對ADPG 存在明顯偏好,因此,該研究團隊分別解析了MtrOtsA 與ADPG 或GDPG 結合的二元復合物晶體,對比分析二者結構發現:ADPG 與MtrOtsA 氨基酸殘基Val363 和Leu319 均可形成氫鍵,而GDPG 僅可與MtrOtsA 的Val363 殘基形成氫鍵,這表明與GDPG 相比,APDG 可與MtrOtsA 形成更加緊密的相互作用,這為MtrOtsA 的底物偏好性提供了有力的結構生物學證據。此外,該研究還通過關鍵氨基酸殘基突變等研究證明:MtrOtsA 對催化底物的偏好性主要是由Leu319、Val363 和Glu367 這3 個殘基介導的。可見,不同物種海藻糖-6-磷酸合成酶的NDPG 結合口袋大致是保守的,僅在與堿基結合位點處表現出細微差異,這可能也解釋了不同物種海藻糖-6-磷酸合成酶對供體底物存在著不同偏好的原因。
1.1.4 海藻糖-6-磷酸合成酶與糖基受體底物G6P 的結合模式分析 海藻糖-6-磷酸合成酶的糖基受體底物G6P 大部分結合于該酶N-端Rossmann結構域,可以通過氫鍵作用力、疏水作用力、靜電作用力等與該酶催化活性中心的氨基酸殘基產生相互作用 (圖5)。
研究發現,海藻糖-6-磷酸合成酶N-端結構域內部存在著一個正電結合口袋,底物G6P 的磷酸酯基可通過靜電相互作用與這一口袋產生結合。如EcOtsA 中這一正電口袋是由Arg9 和Arg300 兩個殘基形成的[14],分別對應著MtrOtsA 中Arg18和Arg324 以及MoTPS1 中Arg22 和Arg327[40-41]組成的各自正電結合口袋。由此可以看出:參與該靜電特征結合口袋形成的氨基酸殘基在不同生物體內是具有高度保守性的。除上述靜電相互作用外,G6P 磷酸酯基還可以與海藻糖-6-磷酸合成酶N-末端保守的酪氨酸殘基以氫鍵產生相互作用,進一步增強了底物與酶活性中心的親和力。
受體底物G6P 的葡萄糖基被由若干個氨基酸殘基形成的疏水口袋包圍,同時也可通過形成氫鍵作用與海藻糖-6-磷酸合成酶活性中心的氨基酸殘基產生相互作用。經比對分析發現:與G6P 葡萄糖基產生相互作用的氨基酸殘基雖然表現出細微的物種特異性,但大部分氨基酸殘基在不同物種間仍是相對保守的。Mendes 等[40]通過氨基酸序列同源性分析發現:MtrOtsA 中與G6P 葡萄糖基結合的關鍵殘基Asp144 等在真菌TPS1 和細菌OtsA 中也是保守的,而殘基Gln146 僅在耐熱分枝桿菌及其近緣物種中是保守的,該殘基對應其他物種海藻糖-6-磷酸合成酶中保守的組氨酸殘基。Miao 等[42]也證實,CaTPS1 中與G6P 葡萄糖基相互作用的氨基酸殘基Asp143 和His145 等在物種間也是高度保守的。
分析與G6P 結合的海藻糖-6-磷酸合成酶三維空間結構,除可得到“配體-受體”相互作用模式外,還可以為進一步闡明酶催化機制提供科學性證據。目前,已有糖基轉移酶兩種催化機制被闡明,分別為保留催化機制和反轉催化機制。海藻糖-6-磷酸合成酶則采取保留催化機制,即在催化葡萄糖基轉移過程中不發生異頭碳原子構型的改變,且該催化機制在不同生物體內也是相當保守的[30,37]。Gibson 等[14]通過解析以G6P 和UDP 為配體的EcOtsA 三維結構發現:EcOtsA 內部包圍G6P 葡萄糖基部分的疏水結合口袋中存在著兩個保守氨基酸殘基:Trp85 和Ile155,二者形成的疏水環境僅可使呈α-構型的葡萄糖O1 羥基處于某一特定位置,以便于其親核攻擊相應的供體葡萄糖基,最終只能形成α,α-1,1 糖苷鍵。該結論首次以海藻糖-6-磷酸合成酶三維結構為依據佐證了海藻糖-6-磷酸合成酶是采取保留催化機制的觀點。此外,Gibson 等[14,36]還研究報道了在EcOtsA的N-端結構域存在著保守殘基Trp40,該殘基可能是發揮著G6P 進出蛋白活性空腔的關鍵。即當G6P 進入EcOtsA 活性中心后,其關閉該位點,而催化完成后,則開啟該位點以釋放產物。Wang 等[41]通過疊合分析MoTPS1 單體、二元復合物 (MoTPS1+UDPG) 和三元復合物 (MoTPS1+T6P+UDP) 結構,進一步對G6P 進出海藻糖-6-磷酸合成酶的門控機制進行了闡釋。該研究指出,MoTPS1 單體與其二元復合物結構類似,但相比于單體和二元復合物,其三元復合物構象卻更為緊湊,基于此提出了海藻糖-6-磷酸合成酶催化“開-關”模型,即:UDPG 首先進入TPS1 催化中心并與之結合,但這一結合并未改變TPS1 分子整體構象,也就是此時TPS1 仍呈現“開放”狀態;當G6P 隨后進入TPS1 并結合至其位于N-末端催化中心時,會導致TPS1 構象發生改變,隨即轉入“關閉”狀態,在此狀態下G6P 與UDPG 相互靠近,并完成糖基轉移的親核反應;反應完成后,TPS1 構象又發生改變再度呈現為“開放”狀態,分別釋放反應產物T6P 和UDP,完成這一催化反應。此外,分析CaTPS1 和MtrOtsA 等多元復合物三維結構也發現了與MoTPS1 類似的構象變換特點,這進一步支持了海藻糖-6-磷酸合成酶在催化時存在門控“開-關”機制的假設。
綜上所述,不同物種海藻糖-6-磷酸合成酶的三維結構雖略有差異,但總體結構仍具有較高相似性,通過對不同生物海藻糖-6-磷酸合成酶蛋白序列進行比對 (圖6) 發現:該酶底物結合位點的關鍵氨基酸殘基同源性較高,表明酶活性中心底物結合口袋構象相對保守,這為基于海藻糖-6-磷酸合成酶設計廣譜性新農藥提供了重要依據。此外,海藻糖-6-磷酸合成酶與底物結合的關鍵氨基酸殘基和關鍵作用力的解析以及酶催化機制的闡明也為基于靶標結構的新農藥分子設計開拓了新思路。
1.2 海藻糖-6-磷酸磷酸酯酶晶體結構及其與底物結合模式分析
除海藻糖-6-磷酸合成酶外,海藻糖-6-磷酸磷酸酯酶作為海藻糖合成酶中另一個重要功能域,其位于海藻糖合成途徑下游,主要負責使T6P 去磷酸化最終生成海藻糖 (圖1)。在生物體內,磷酸酯酶依賴鎂離子催化磷酸根水解反應,是生物化學反應和能量代謝的重要參與者。生物體內磷酸酯酶種類眾多,但其在高級結構上卻表現出較高相似性。因此,學者們將具有類似結構的磷酸酯酶劃分為鹵酸脫鹵酶 (HAD) 超家族,這些酶均具有兩個結構域,其中核心結構域呈典型的Rossmann折疊,另一個結構域為帽型,這兩個結構域在不同生物體內是高度保守的,但相比較帽型結構域大小往往會存在一定差異[44]。
本文介紹的海藻糖-6-磷酸磷酸酯酶就隸屬于HAD 超家族,目前已有近10 種生物的海藻糖-6-磷酸磷酸酯酶晶體結構已陸續被解析 (表3),這些蛋白晶體結構均具有HAD 超家族典型結構特征。Rao 等[45]于2006 年首次報道了嗜酸熱原體菌Thermoplasma acidophilum 海藻糖-6-磷酸磷酸酯酶TaOtsB 晶體結構,其核心結構域是一個經修飾的Rossmann 折疊結構域,它是由8 個β 折疊片層和兩側5 個α 螺旋構成的,其中β4 和β5 在β3 處形成了明顯的發夾結構。而其帽型結構域包含4 個β 折疊片和2 個α 螺旋,主要是通過兩條蛋白鉸鏈與其核心結構域相連的,TaOtsB 活性中心主要位于Rossmann 折疊結構域和帽結構域之間界面處,由3 個保守氨基酸序列構成。Cross 等[46]報道了銅綠假單胞菌Pseudomonas aeruginosa PaOtsB蛋白結構,通過解析該結構發現:PaOtsB 結構域具有與TaOtsB 幾乎完全相同的折疊模式,僅β-發夾處存在著細微差異,其PaOtsB 核心結構域的β-發夾是由β2 鏈和β3 鏈折疊而成的。細菌假伯克霍爾德氏菌Burkholderia pseudomallei 的海藻糖-6-磷酸磷酸酯酶BpOtsB 同樣也具有HAD 超家族酶保守結構,其核心結構域同樣為8β5α 型,其β4鏈與其他β 折疊片反平行,并無類似于TaOtsB中的發夾結構,其C2B 帽型結構域同樣是由4 個β 折疊片和2 個α 螺旋構成的[47]。
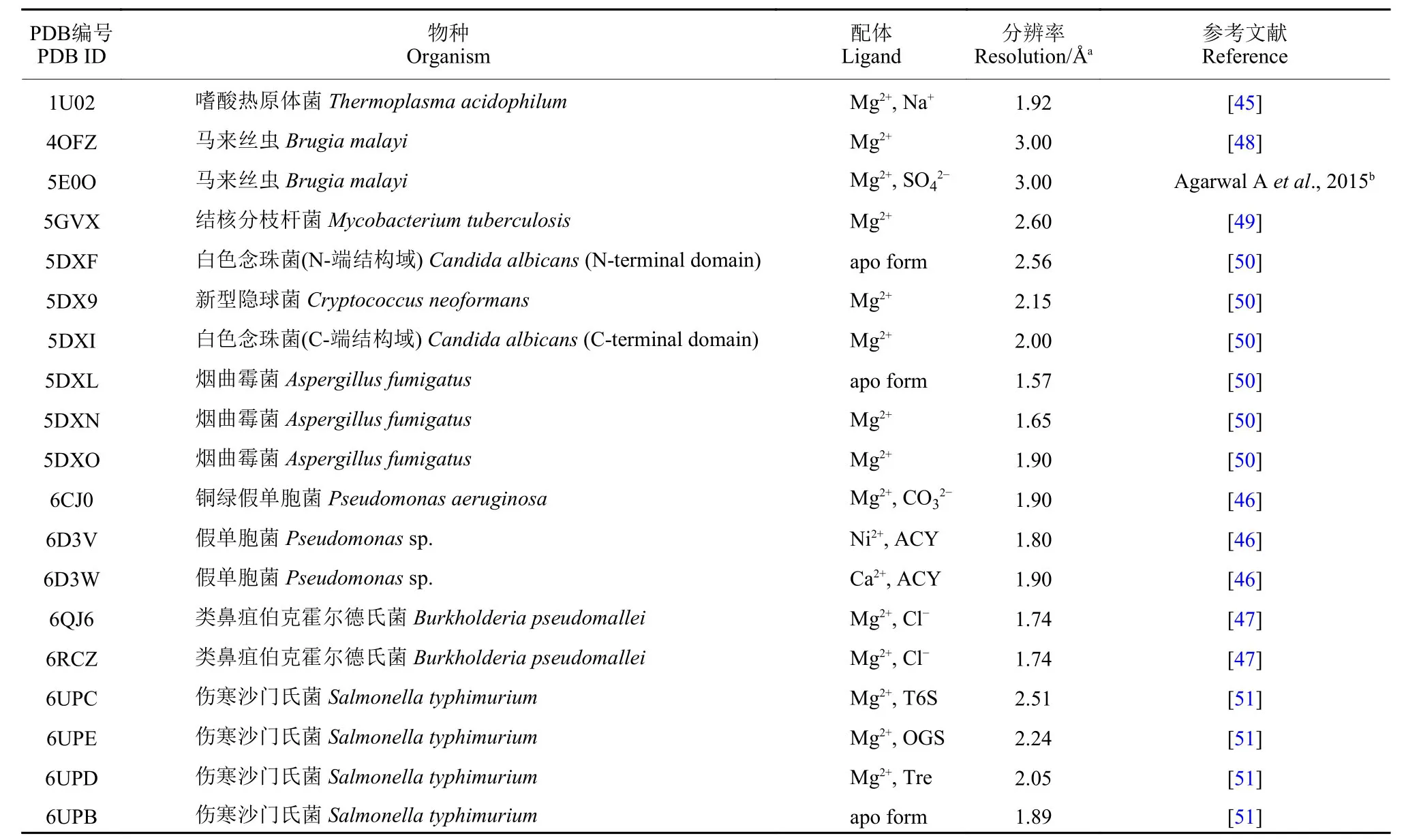
表3 目前已報道的不同海藻糖-6-磷酸磷酸酯酶晶體結構信息Table3 The reported crystal structures of different trehalose-6-phosphate phospholipase
不同生物的海藻糖-6-磷酸磷酸酯酶總體結構是呈現較高相似性的,但往往也存在物種間差異性。如馬來絲蟲 Brugia malayi TPP (BmTPP) 蛋白除擁有HAD 超家族酶普遍存在的核心結構域和帽型結構域外,還具有一個由3 股螺旋束構成的N-端結構域[48]。來自細菌Mycobacterium tuberculosis 和真菌C. albicans 的海藻糖-6-磷酸磷酸酯酶晶體結構顯示:這兩個海藻糖-6-磷酸磷酸酯酶空間結構也是由核心結構域、帽型和N-端結構域構成的,在3 個結構域之間通過一段或幾段多肽鏈進行偶聯。與馬來絲蟲BmTPP 結構不同的是,結核分枝桿菌MtbOtsB 和白色念珠菌CaTPS2 的N-端結構域空間折疊方式更為復雜,且均為典型的Rossmann結構域。Miao 等通過結構比對發現,CaTPS2 的N-端結構域與大腸桿菌EcOtsA 結構類似,但卻缺少EcOtsA 中與底物結合的關鍵氨基酸殘基,如Arg9、Tyr76 和Arg300 等[49-50]。因此,表明CaTPS2的N-端結構域不具有合成T6P 的功能,推測該結構域或許可與另一亞基CaTPS1 互作,并在TPS1折疊和催化過程中發揮著一定功能,但該假說至今尚未得到實驗證實。
不同生物海藻糖-6-磷酸磷酸酯酶的晶體結構也揭示了其相當保守的底物結合模式。Rao 等[45]通過模擬TaOtsB 與其底物T6P 結合,發現Ile16、Thr45、Lys111、His118、Lys149、Glu153 和Lys161是TaOtsB 與T6P 產生互作的關鍵氨基酸殘基,且上述氨基酸殘基在不同物種的海藻糖-6-磷酸磷酸酯酶中保守性極高。Harvey 等[51]分別解析了傷寒桿菌Salmonella typhimurium 的StOtsB 與第1 代海藻糖-6-磷酸磷酸酯酶抑制劑海藻糖-6-硫酸 (T6S)和1-(4-己基苯氧基)-葡萄糖-6-硫酸 (OGS) 復合蛋白晶體結構,并報道了StOtsB 與底物結合方式,發現StOtsB 中Glu123、Lys125 和Glu167 等殘基是與T6S 及OGS 結合的關鍵殘基,且保守度極高。這些關鍵氨基酸殘基的解析,為以海藻糖-6-磷酸磷酸酯酶為靶標的新一代抑制劑合成和先導化合物改造提供了重要依據,也為靶向海藻糖-6-磷酸磷酸酯酶的廣譜性農藥設計與開發提供了有力參考。
2 海藻糖合成酶抑制劑
在基于靶標開展新農藥分子設計與研發過程中,已知靶標酶抑制劑化學結構既可作為其他潛在酶抑制劑篩選的依據,也可為先導化合物結構優化與改造提供思路。截至目前,國內外先后報道了10 余個海藻糖-6-磷酸合成酶抑制劑結構 (表4)和若干個海藻糖-6-磷酸磷酸酯酶抑制劑結構 (表5)。根據這些抑制劑分子發現途徑不同,可將其劃分為3 種類型:其一,根據靶標酶天然底物結構進行改造得到的底物類似物;其二,靶標酶的負反饋調節抑制劑及其類似物;其三,基于靶標酶結構開展虛擬篩選或高通量篩選獲得的酶競爭性結合抑制劑。應當注意的是,雖已有一些海藻糖合成酶抑制劑研究報道,但具有較好農用酶抑制活性的抑制劑卻寥寥無幾。因此,筆者按照上述分類方式分別對海藻糖合成酶中兩個重要功能域——海藻糖-6-磷酸合成酶和海藻糖-6-磷酸磷酸酯酶的抑制劑結構和活性進行較為全面綜述,期望為靶向這兩種酶的農用抑制劑合理設計和生物活性評價提供參考。
2.1 海藻糖-6-磷酸合成酶抑制劑
2.1.1 負反饋抑制劑及其類似物 負反饋抑制是生物體內普遍存在的酶活調控方式,是指酶催化產物作為抑制劑反過來調控酶活性,廣義也可指系列反應中下游產物對上游催化酶活性的抑制。通常,在一定范圍內,產物濃度與負反饋抑制效果間存在著明顯正相關性[52]。
T6P 是海藻糖-6-磷酸合成酶催化產物,其被稱為該酶的負反饋抑制劑,且其發揮酶負反饋抑制效應已被闡明。Magalh?es 等[52]發現,在體外條件下,T6P 會顯著抑制釀酒酵母 Saccharomyces cerevisiae ScTPS1、白色念珠菌CaTPS1 和熱帶念珠菌Candida tropicalis CtTPS1 活性:當T6P 濃度為125 μmol/L 時,CtTPS1 活性降低約50%,CaTPS1 活性降低約60%。結合酶促動力學分析結果表明:T6P 是海藻糖-6-磷酸合成酶的有效非競爭性抑制劑。此后,有研究者通過同源模建構建了ScTPS1 蛋白模型,并將經能量優化的T6P 分子與該模型進行兩次對接。結果顯示:T6P 分子結合于ScTPS1 中一固定活性腔內,并與高度保守的氨基酸殘基Tyr40、Ala41、Met42 和Phe372 通過氫鍵產生相互作用[53],進一步揭示了T6P 可作為海藻糖-6-磷酸合成酶抑制劑用于新藥研發,但T6P 分子由于其磷酸基團的存在,使其不容易穿越細胞膜被生物體吸收。
海藻糖是生物體內海藻糖合成代謝的終產物,經相關研究表明,在海藻糖-6-磷酸合成酶活性負反饋調控中,海藻糖也發揮了重要功能。約半世紀前,Murphy 等首次報道了在蠶蛾體內海藻糖對其TPS 活性存在明顯負反饋調控功能[54],但此后就鮮有關于海藻糖參與調控海藻糖-6-磷酸合成酶活性的報道。直至2017 年,Oide等[55]通過對離體谷氨酸棒狀桿菌Corynebacterium glutamicum CgTPS1 蛋白進行酶促動力學分析發現:對CgTPS1而言,海藻糖是其供體底物UDPG的競爭性抑制劑,是其受體底物G6P 的非競爭性抑制劑,海藻糖對CgTPS1 酶活性負反饋調節占主導地位,而非T6P。Mendes 等[40]也證實:在生理條件下,海藻糖對耐熱分枝桿菌MtrOtsA 活性存在一定抑制效果,且其半抑制濃度約為24 mmol/L。
井岡霉素A (validamycin A) 是一種產量大且應用廣的微生物源農藥,對小麥、水稻紋枯病表現出較好防效,其水解產物井岡羥胺A (validoxylamine A) 作為海藻糖類似物,早已被證實可作為海藻糖酶有效抑制劑[56-57]。然而,近些年研究表明:井岡羥胺A 可作為海藻糖-6-磷酸合成酶的過渡態抑制
劑[38,42],且白色念珠菌CaTPS1 和煙曲霉菌AfTPS1與井岡羥胺A 結合的復合物蛋白結構已被解析[30]。Errey 等[38]基于井岡羥胺A 結構,合成了6′-井岡羥胺A 磷酸 (6′-validoxylamine A phosphate),該化合物對EcOtsA 表現出較強抑制效果,其IC50值約為5.3 mmol/L,且進一步研究發現:在UDP存在條件下,井岡羥胺A-6′-磷酸對EcOtsA 的抑制效應會明顯增強。

表4 目前已報道的不同海藻糖-6-磷酸合成酶抑制劑Table4 The reported inhibitors of different trehalose-6-phosphate synthetase

續表 4Table4 (Continued)
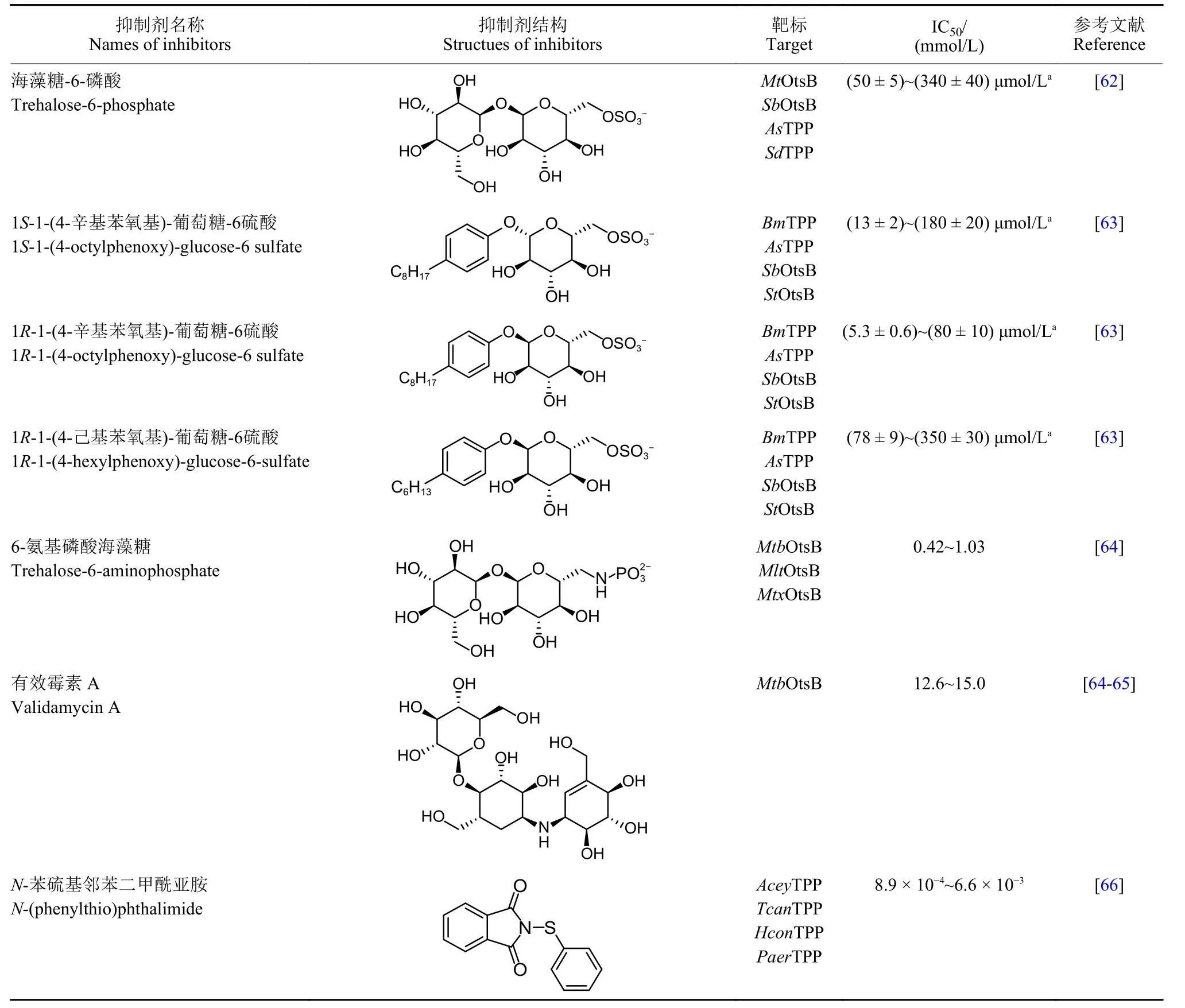
表5 目前已報道的不同海藻糖-6-磷酸磷酸酯酶抑制劑Table5 The reported inhibitors of different trehalose-6-phosphate phosphatase
除上述海藻糖代謝相關產物可對海藻糖-6-磷酸合成酶活性存在負反饋抑制外,與海藻糖代謝密切相關的其他糖代謝中間產物也可對該酶活性產生負反饋抑制效應。Mendes 等[40]通過測試若干糖酵解中間物對MtrOtsA 抑制活性研究發現:2-磷酸甘油酸 (2PG) 和酮戊二酸 (2OG) 可作為別構調節劑顯著抑制MtrOtsA 活性,其IC50值分別為2.3 mmol/L 和1.8 mmol/L。
由此看來,雙糖類和擬雙糖類抑制劑在已報道的海藻糖-6-磷酸合成酶抑制劑中占比最大,且大部分抑制劑具有較出色的酶抑制活性。因此,在未來設計海藻糖-6-磷酸合成酶抑制劑時,可考慮引入糖環或雙糖結構,期望有利于新合成化合物的酶抑制效果。同時應注意化合物相關理化性質并對其結構進行合理優化,以提高其在生物體內的吸收和利用率,使其既可達到良好的酶活性抑制效果,又可表現出優異的殺滅有害生物活性。
2.1.2 競爭性結合抑制劑 除生物體內存在的某些天然化合物可作為海藻糖-6-磷酸合成酶活性負反饋抑制劑外,通過應用計算機和分子生物學技術開展海藻糖-6-磷酸合成酶抑制劑的虛擬篩選或高通量篩選也已成為新型糖代謝相關酶抑制劑發現的另一重要途徑,且篩選得到的化合物大多為酶的競爭性結合抑制劑。
Xue 等[58]通過同源模建方法構建得到了稻瘟病菌MoTPS1 三維結構模型,通過將該結構模型與MLSMR 化合物數據庫中40 萬個分子開展批量分子對接,篩選得到了與MoTPS1 親和力最強的化合物2 478 993,并以其為母體進行結構優化,通過引入羰基得到了先導化合物Lead 25,該化合物既表現出較高的MoTPS1 親和力 (親和自由能為-13.8 kcal/mol,即 -57.76 kJ/mol),又具有較強水溶性。此外,作者還發現:與海藻糖-6-磷酸合成酶的天然底物UDPG 和G6P 相比,NADPH 與MoTPS1 具有更強的親和力 (UDPG 親和自由能:-10.4 kcal/mol,即 -43.53 kJ/mol;G6P 親和自由能:-7.0 kcal/mol,即 -29.30 kJ/mol;NADPH 親和自由能:-11.3 kca/lmol,即 -47.30 kJ/mol),這表明NADPH 也可作為海藻糖-6-磷酸合成酶的潛在抑制劑進行進一步開發。該研究是針對海藻糖-6-磷酸合成酶篩選和開發農用殺菌劑的首次成功試驗,這為將來基于MoTPS1 結構開展新抑制劑的發現提供了借鑒和經驗。
Verma[59]使用熱位移分析法對耐熱分枝桿菌MtrOtsA 抑制劑進行了Ⅰ、Ⅱ兩種類型的高通量篩選,其中Ⅰ型篩選得到15 個MtrOtsA 潛在抑制劑分子,Ⅱ型篩選得到7 個在腺苷二磷酸 (ADP)存在條件下的MtrOtsA 潛在抑制劑分子,隨后分別測定了兩類型篩選所得化合物的IC50值。結果顯示:5-(2-噻吩基) 煙酸對MtrOtsA 抑制效果最佳,其IC50值約為0.5 mmol/L,在ADP 存在條件下,4-羥基-6-(三氟甲基)-3-喹啉羧酸為抑制活性最強的MtrOtsA 抑制劑,其IC50值約為0.53 mmol/L。值得注意的是:經Ⅰ、Ⅱ兩類型篩選得到一共同分子——5-氨基吲哚,該分子以0.65 mmol/L 的IC50值表現出顯著的抑制MtrOtsA 活性,且化合物結構簡單,改造潛力巨大。因此,在今后針對海藻糖-6-磷酸合成酶抑制劑進行結構優化時,應對5-氨基吲哚這一結構片段給予充分關注。
2017 年,來自杜克大學的Perfect 等[60]利用UDP 熒光偏振技術篩選得到了若干個白色念珠菌CaTPS1 和煙曲霉菌AfTPS1 抑制劑,其中克羅散泰 (Closanthal) 在體外條件下對隱球菌存在著顯著抑制效應,其最小抑菌質量濃度為1 mg/L,但在體內條件下,克羅散泰并沒有發揮良好的抑菌效果,這或許與藥物在生物體內復雜的吸收和代謝機制有關。
Kern 等[61]通過克隆并表達黑腹果蠅Drosophila melanogaster 和貓蚤TPS 基因進行了首次靶向昆蟲TPS 的抑制劑高通量篩選嘗試,并得到了一系列對黑腹果蠅DmTPS 和貓蚤CfTPS有顯著抑制效果的化合物,結構如表4 所示。其中2,6-二氨基-4-(3,4-二氯苯基)-3,5-二氰基-4-H-噻喃對DmTPS的抑制效果最佳,其IC50值僅為0.2 mmol/L,且以該結構為骨架合成的若干化合物對DmTPS 和CfTPS 也表現出優良抑制效果。此外,2,6-二氨基-4-(3-環己烯基)-3,5-二氰基-4-H-噻喃、2,6-二氨基-4-(2-硝基噻吩基)-3,5-二氰基-4-H-噻喃等結構及其衍生物也對昆蟲TPS 存在一定抑制效應,這些化合物的IC50值范圍分布在1.4~6.2 mmol/L 之間。這一研究為基于海藻糖-6-磷酸合成酶開發新型農用殺蟲劑奠定了基礎。
虛擬篩選和高通量篩選是酶抑制劑篩選和先導化合物發現的高效途徑,許多研究者通過分子對接、藥效團篩選、等溫量熱、熒光偏振和熒光熱位移分析等方法均在短時間內實現了從大規模化合物庫中篩選獲得潛在的海藻糖-6-磷酸合成酶抑制劑,這提示我們在今后開展酶抑制劑篩選工作時,要注意整合優化篩選模式,綜合運用多種篩選方法,有望更加高效地篩選出有更優活性的新型酶抑制劑。
2.2 海藻糖-6-磷酸磷酸酯酶抑制劑
有關海藻糖-6-磷酸磷酸酯酶抑制劑的研究于近年來剛剛起步,相關研究較少,但也報道了一些對海藻糖-6-磷酸磷酸酯酶有較好抑制效果的活性化合物,其中絕大部分為該酶天然底物T6P 類似物,僅有一種酶抑制劑為通過高通量篩選方法所得。
2.2.1 底物T6P 及其類似物 2017 年,Liu 等[62]首次嘗試了海藻糖-6-磷酸磷酸酯酶抑制劑的合成,他們基于底物T6P 結構設計并合成了4 種底物類似物,這4 種底物類似物均保留了T6P 雙糖結構,且都含有一個C6 位取代的類似磷酸根負電基團。然而,在上述4 種化合物中,僅海藻糖-6-硫酸 (T6S) 表現出對不同生物TPP 或OtsB 的一定抑制活性,經測定其Ki值主要分布在 (50 ± 5) ~ (340 ±40) μmol/L 之間,且具有較天然底物T6P 更強的結合活性 (T6P 的Km值為 (230 ± 70) ~ (690 ± 70)μmol/L)。
隨后,該團隊又開展了對T6S 結構優化工作,以苯環代替T6S 中葡萄糖環,并以此為母體合成了一系列化合物,其中1R-1-(4-辛基苯氧基)-葡萄糖-6-硫酸對不同生物海藻糖-6-磷酸磷酸酯酶的抑制活性最高,其Ki值僅為 (5.3 ± 0.6) ~ (80 ±10) μmol/L 之間。此外,1S-1-(4-辛基苯氧基) -葡萄糖-6-硫酸和1-(4-己基苯氧基)-葡萄糖-6 硫酸也表現出較好的酶抑制活性,二者Ki值分別為 (13 ± 2) ~(180 ± 20) μmol/L 和 (78 ± 9) ~ (350 ± 30) μmol/L[63],進一步構效關系分析發現,含有較長烴鏈且為α 異頭型的T6S 衍生物普遍表現出較高的酶抑制活性。
2019 年,Kapil 等[64]也進行了以T6P 為母體的酶抑制劑結構改造,其合成的化合物6-氨基磷酸海藻糖對3 種分枝桿菌OtsB 均有較好抑制效果,其IC50值在0.42~1.03 mmol/L 之間。同時,鑒于有研究報道,有效霉素A 可作為EcOtsB[65]的有效抑制劑,因此上述研究中[64]也測定了有效霉素A 對結核分枝桿菌OtsB 的抑制活性,結果顯示:有效霉素A 雖可對結核桿菌OtsB 產生一定抑制效應,但其抑制效果并不樂觀,其IC50值為12.6~15.0 mmol/L。
2.2.2 競爭性結合抑制劑 海藻糖-6-磷酸磷酸酯酶是近年來新發現的殺蟲劑、殺菌劑候選靶標,具有較大開發潛力,但目前有關該酶抑制劑的篩選工作研究仍較少。Cross 等[66]制定了兩步篩選策略,從4 個化合物文庫 (Natural products, Pathogen Box, CSIRO synthetic library 和Open Scaffolds Library) 的 5 000 余種化合物中篩選得到了N-苯硫基鄰苯二甲酰亞胺,如表5 所示。該化合物對錫蘭鉤口線蟲Ancylostoma ceylanicum、犬弓蛔蟲Toxocara canis 等4 種線蟲的TPP 均表現出強抑制性,其IC50值低至0.89~6.6 μmol/L。且經過體外試驗研究發現:該化合物會使線蟲成蟲成活率和幼蟲遷移率明顯下降,證明該化合物既表現出很高的酶抑制活性,又具有良好的殺蟲活性和驅蟲活性,但其對細菌OtsB 幾乎無抑制效果,這可能是由于細菌與線蟲海藻糖-6-磷酸磷酸酯酶在空間結構上存在差異所致。
3 結論與展望
當前,利用農藥進行化學防治是有害生物綜合治理的主要方法,但農藥過量和不合理使用導致了藥劑防效降低、有害生物抗藥性出現以及生態環境被破壞等一系列問題,甚至嚴重威脅了非靶標生物生存和正常生理過程,因此,探索新農藥靶標并基于新靶標進行合理藥物設計是解決這一問題的主要手段。海藻糖在病原真菌、細菌侵染及昆蟲生長發育等生理過程中發揮著重要功能,更為重要的是與海藻糖代謝相關酶在人等哺乳動物體內缺失,這表明海藻糖合成酶可作為抗病原生物藥物開發的潛在靶標,用于非靶標安全型新農藥的開發。本文主要綜述了參與海藻糖合成的海藻糖-6-磷酸合成酶和海藻糖-6-磷酸磷酸酯酶的晶體結構特征、酶與底物結合的關鍵氨基酸殘基,為基于靶標的新農藥分子設計提供結構依據,隨后對已報道的酶抑制劑化學結構、生物活性及構效關系進行評述,以期對今后靶向海藻糖合成相關酶抑制劑的篩選、先導結構的發現與優化提供有意義指導。
雖然有關海藻糖合成酶結構及其抑制劑研究已取得一定進展,但仍存在許多問題,如海藻糖合成酶晶體結構較少,且尚無昆蟲海藻糖合成酶晶體結構報道;現有酶抑制劑結構較為復雜且單一,已報道酶抑制劑雖具有較好離體活性,但其活體活性較差或鮮有報道。綜上所述,未來研究應聚焦解析更多農業有害生物的海藻糖合成相關酶晶體結構,尤其是針對昆蟲海藻糖合成酶結構的獲取,以實現基于酶結構進行新農藥分子的合理設計。在以海藻糖合成酶為靶標的抑制劑篩選過程中,應不斷優化改進以實現多種篩選方法融合,預期發現酶抑制活性高的先導化合物,并進行進一步結構改造。隨著相關研究的不斷深入,可以有力推動以海藻糖合成酶為靶標的新農藥設計與開發,為現代農業有害生物防治提供全新解決方案。
猜你喜歡
小獼猴智力畫刊(2023年4期)2023-04-23 08:49:58
哲學評論(2021年2期)2021-08-22 01:53:34
中華詩詞(2019年7期)2019-11-25 01:43:04
模具制造(2019年3期)2019-06-06 02:10:54
中學生數理化·高一版(2018年1期)2018-02-10 05:20:03
影視與戲劇評論(2016年0期)2016-11-23 05:26:01
七彩語文·寫字與書法(2016年7期)2016-07-28 21:40:22
七彩語文·寫字與書法(2016年6期)2016-07-15 19:36:34
人間(2015年21期)2015-03-11 15:23:21
現代企業(2015年9期)2015-02-28 18:56:50
