薄殼山核桃SCAR標記開發及其在品種間的多態性
2021-04-10 06:12:58,,,,
經濟林研究 2021年1期
,,,,
(1.浙江省林業科學研究院,浙江 杭州 310023;2.森林食品資源利用與質量控制國家林業局重點實驗室,浙江 杭州 310023)
薄殼山核桃Carya illinoinensis(Wangenh.) K.Koch是胡桃科山核桃屬植物,因其性狀優良和核仁營養價值高,在世界五大洲被廣泛引種栽培,是重要的世界性干果和油料作物[1-2]。薄殼山核桃是雌雄同株的風媒花異交植物,按照雌雄花成熟期的先后,大致可分為雄先型和雌先型2 種類型[3]。不同品種在不同氣候條件下會存在花期不遇的問題,這是影響薄殼山核桃穩產和高產的重要原因之一。美國是薄殼山核桃的原產地之一,目前也是薄殼山核桃的中心產區,其產量占全球產量的85%以上。美國現有1 000 多品種,在建園時往往可以根據當地的氣候特點進行有效的雜交配置,壯年林干果產量可達1 500 kg/hm2以上。雖然我國引種薄殼山核桃已有100 多年的歷史,但是目前生產上常用的品種僅幾十個。多年的生產實踐結果證明我國不少地區是薄殼山核桃的適生區,但是較多薄殼山核桃產區面臨產量低下和產量不穩定的問題,尤其是廣大亞熱帶地區。造成這一現象的原因有多個方面,比如實際使用的品種較為有限,再加上引種時存在錯誤以及一些雜交后代定名混亂,難以進行有效的親本選擇和親緣關系分析,更難以根據當地的氣候特點進行有效的雜交配置。
目前,用于薄殼山核桃品種鑒定的分子標記方法主要有RAPD[4-5]、SSR[6-9]、SRAP[10]、ISSR[11-12]和SNP[13]等。一般來說,SSR 和SNP 等標記須進行大量基因組或轉錄組的測序和分析[13-14],得到穩定性和多態性較好的標記的成本較高。在利用SSR引物對薄殼山核桃進行品種鑒定時,重復性檢測中也存在一些問題[7],這主要是因為:一方面薄殼山核桃高度雜合,基因組大且有較多重復,導致SSR 引物存在一定的非特異性;另一方面,SSR 和SNP 這類標記對檢測的要求較高,且檢測識別時存在一定的人為干擾。隨著測序技術的進步以及越來越多的薄殼山核桃基因組數據的公布,利用品種間基因組數據的差異挖掘序列特異性標記(sequence characterized amplified region,SCAR)是一個提高分子標記的穩定性和特異性的辦法。本研究中通過對不同薄殼山核桃品種葉片DNA 進行簡單基因組測序,尋找品種特異性的SCAR 標記,旨在針對生產上常用品種從分子水平上開發出穩定、特異的DNA 指紋標記,為輔助實現薄殼山核桃品種準確快速鑒定和親緣關系分析提供參考。
1 材料與方法
1.1 試驗材料
薄殼山核桃樣品采集于建德薄殼山核桃基地,地理位置為119°32.86′E、29°33.76′N,海拔97.5 m。5月上旬,采摘植株新鮮嫩葉,冰浴運輸,當天保存于-20 ℃冰箱中備用。所涉及的品種見表1。其中,Ind23 和Ind24 為不同來源的同一品種,Ind23 為20 世紀80年代從美國德克薩斯州美國山核桃試驗站引進,Ind24 為1999年從美國國家無性系種質資源庫(National Clonal Germplasm Repository)引進。“雄先”和“雌先”的判斷是根據不同性別花剛開始成熟的時間,雄花是指花粉開始散粉,雌花是指柱頭出現可授粉狀態[3]。

表1 薄殼山核桃試驗品種及其生理特點Table 1 Experimental varieties and physiological characteristics of C.illinoensis
1.2 試驗方法
1.2.1 DNA 提取
葉片DNA 的提取采用新型快速植物基因組DNA 提取試劑盒(BioTeke,北京)。取各品種葉片樣品0.02 ~0.03 g,剪碎后放入2.0 mL 離心管內,加入直徑3 mm 碳化鎢研磨珠2 顆,用液氮冰凍5 ~10 min,再用MM400 研磨儀(萊馳,德國)30 Hz 研磨2 min,然后按照試劑盒說明書操作。提取好的DNA 經1.0%的瓊脂糖凝膠電泳檢測完整性,然后用NanoDrop2000 超微量分光光度計檢測其質量濃度,置于-20 ℃冰箱備用,使用前將DNA 質量濃度稀釋標定至20 ng/μL。
1.2.2 簡化基因組測序和分析
簡化基因組測序是基于RAD(restriction-siteassociated DNA sequencing)方法進行的。具體步驟:提取高質量DNA,使用多種限制性內切酶對DNA樣品分別進行酶切,根據酶切試驗的結果選擇合適的酶,構建長度為300 ~500 bp 的pair-end 文庫,用Illumina HiSeq PE150 測序文庫,獲得每種樣品的簡化基因組原始數據。
后續分析方法:1)對每種樣品中的Reads1(雙端PE 測序后前端的數據,即含有EcoRI 酶切位點AATTC 的片段)進行比對聚類;2)對每種樣品的序列聚類結果進行內部比對,得到樣品內部各序列位點信息;3)將不同樣品的序列互相進行比對,尋找個體之間堿基差異信息,并根據樣品信息尋找亞群特異的序列位點;4)根據特異序列信息獲得雙端數據進行組裝,得到亞群特異的基因組片段。
具體組裝方法:1)使用cap3 軟件(http://seq.cs.iastate.edu/cap3.html)將Reads1 與Reads2 進 行局部組裝;將特異Taq(unique Tag)與最長的組裝結果進行連接(中間加入5 個“N”),獲得初步組裝結果;2)將某一樣品獲得的組裝Tag 與其他樣品的序列進行blastn 比對,過濾掉相似序列,比對使用的軟件為blastn,參數e-value 為1×10-5。另外,在設計引物前,為了增加引物擴增的有效率,使 用MISA(http://pgrc.ipk-gatersleben.de/misa/)對組裝結果進行SSR 預測,剔除含有SSR 的組裝結果。
使用Vector NTI 9.0 軟件設計引物,各樣品特異片段的引物設計原則:引物長度18 ~22 bp,解鏈溫度56 ~62 ℃,產物長度150 ~300 bp。
1.2.3 SCAR 標記開發
對使用上述引物預測的片段進一步開展blast分析,挑選出與核桃(Juglans regia,現有全基因組數據中與薄殼山核桃最近物種)同源性最高的DNA 序列片段,作為后續SCAR 標記開發的候選序列。對這些序列進行PCR 篩選,挑選出23 個品種中存在多態性的DNA 條帶,切膠回收純化這些條帶,并利用PCR 引物進行兩端測序,測序結果經Vector NTI 9.0 軟件中的ContigExpress 拼接,將重測序得到的序列與之前的基因組測序數據進行比對,補充這些序列中間的未知堿基(這些堿基之前以“NNN”表示),將得到的完整序列作為新的SCAR 標記。
1.2.4 PCR 篩選
常規PCR 試驗所用試劑為BioTeke PCR MasterMix(BioTeke,北京),整個反應體系除引物和模板外,均按照試劑手冊要求操作。在15 μL體系中,引物用量為上游和下游引物(10 μmol/L)各0.6 μL,模板為2 μL。用于直接測序前,反應體系擴大到60 μL,模板為前面15 μL 產物稀釋1 000 倍。混合多對引物進行PCR 反應時,適當調整每對引物的量,至最佳擴增效果,其他條件不變。所有PCR 反應在Genepro Thermal Cycle(Bioer,杭州)PCR 儀上進行,程序:94 ℃ 300 s,95 ℃ 10 s,56 ℃ 50 s,72 ℃ 40 s,共35 輪循環;72 ℃,300 s。擴增產物經1.5%瓊脂糖凝膠電泳檢測,排槍上樣各5 μL,泳道順序對應樣品分別是Ind01,Ind13,Ind02,Ind14……。Marker為DL2000(Takara),條帶大小依次為2 000、1 000、750、500、250、100 bp,上樣量3 μL。EB染色和ChemiDoc(Biorad,北京)拍照。
1.2.5 親緣關系分析
在各品種中,所篩選較為穩定的SCAR 標記的有無,分別以“1”“0”進行記錄,然后使用NTSYS 2.2 軟件建立聚類分支樹狀圖。首先用similarity 程序組中的SimQual 計算形似系數矩陣,然后使用Clustering 程序組中的SHAN 進行計算,聚類方法選用UPGMA,其他參數按默認設置。
2 結果與分析
2.1 RAD 測序和差異分析
選取性狀差異較大的5 個品種的樣品進行Illumina HiSeq PE150 測序,分別是Moore、Nacono、Van Deman、Davis 和Pawnee,測序結果見表2。一般情況下A 與T 數量相等,C 與G 數量相等,(A+T)/(C+G)的值則具有物種特異性。從表2可知,由于該簡化基因組文庫是對酶切片段附近的序列進行測序,所以堿基并不平衡,且頭部5 個堿基為固定的“AATTC”序列,薄殼山核桃(A+T)/(C+G)的值約為1.63。從堿基長度質量指標Q20%和Q30%來看,該文庫堿基質量良好,可用于后續分析。
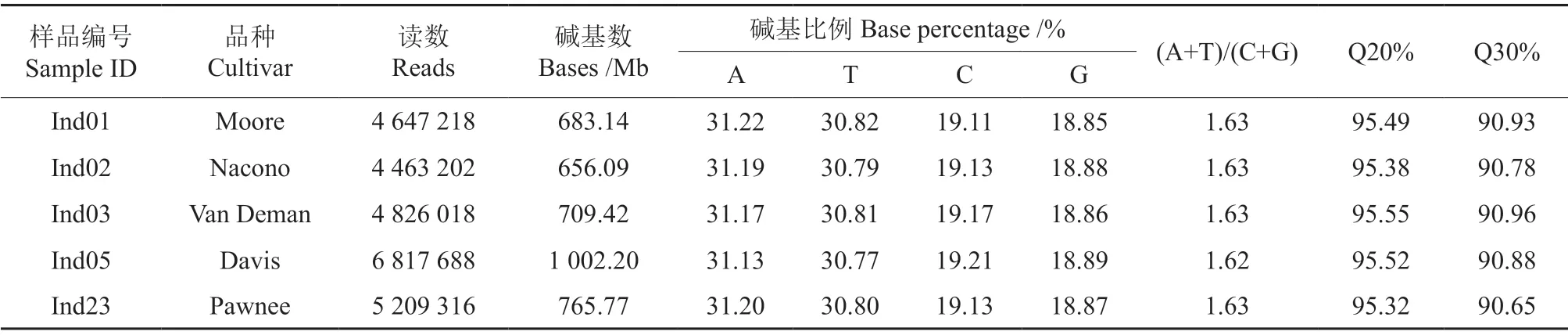
表2 性狀差異較大的5 個薄殼山核桃品種樣品的高通量測序結果Table 2 Deep sequencing results of five C.illinoensis cultivars with different characteristics
測得的數據經初步組裝、blast 分析、精確組裝和比較,得到各品種的特異序列。其中Ind23 特異序列數量最多,達1 080 條,而Ind03 最少,僅為553 條。分析這些片段的二級結構、二聚體形成和堿基分布等,設計了1 133 對引物(表3)。再結合擴增產物與核桃基因組序列的同源性,挑選其中473 對作為最終候選引物。
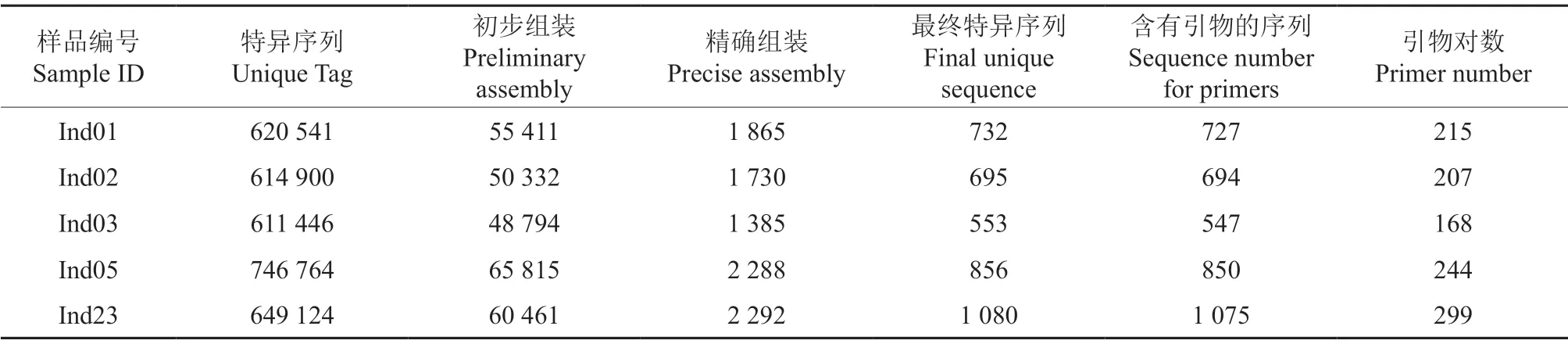
表3 性狀差異較大的5 個薄殼山核桃品種特征序列和引物搜索Table 3 Searching for characteristic sequences and primers of five C.illinoensis cultivars with different characteristics
2.2 PCR 引物篩選結果
PCR 篩選結果表明,絕大部分條帶在各品種間無差異,僅86 對引物在23 個品種中產生多態性,約占引物數量的1/5,其中80 對單一條帶可用于后續分析(圖1)。

圖1 80 對SCAR 引物在23 個薄殼山核桃品種中的多態性分布Fig.1 The polymorphism distribution of 80 SCAR markers among 23 cultivars
2.3 用于品種鑒定的SCAR 引物及其組合
所開發SCAR 標記,大多數在多個品種中僅產生1 個條帶,其中有極少數可以特異性指示單一品種(圖2)。但是,也有少量引物在各品種中產生的條帶數量存在差異(圖3),這些較為復雜的多態性有待深入研究。將產生多態性的這些單一條帶進行組合,獲得一系列可用于品種鑒定的引物組合(表4),這些引物的具體信息見表5。

圖2 薄殼山核桃品種特異性SCAR 標記引物對及單一條帶Fig.2 Cultivar-specific primer pairs and their unique bands of SCAR markers
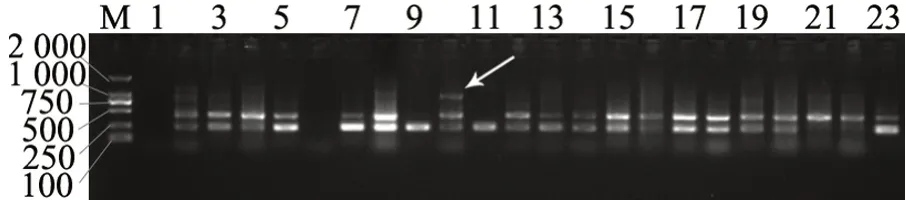
圖3 薄殼山核桃某些SCAR 標記引物的多態性Fig.3 Polymorphism of some scar markers in C.illinoensis
2.4 SCAR 標記用于品種親緣關系的分析
利用80 對引物在各品種間的多態性,進行24個薄殼山核桃品種的聚類分析,結果如圖4所示。由圖4可見,聚類結果與實際的親緣關系有部分一致性,但也存在不一致的地方。例如,來自不同產地的Ind23 和Ind24 同為Pawnee 品種,聚在一起;Ind07(Choctaw)是Success×Mahan 的后代,Ind13(Dependable)是Jewett×Success 的后代,二者聚在一起,可能與其有較近的親緣關系有關;Ind10(Barton)是Moore×Success 的后代,與Ind01(Moore)聚在一起。但是,Ind04(Forkert)是Success×Schley 的雜交后代,Ind07(Choctaw)是Success×Mahan 的雜交后代,同為Success 的雜交后代,二者在聚類關系上卻相差較遠,這可能是其遺傳組合的不同造成的,也可能是因為所篩選SCAR 標記較為有限,在分析親緣關系方面尚存在一定的局限性。
3 結論與討論
因為分子標記能夠從DNA 水平反映品種遺傳的本質,成為品種鑒定最為重要的依據之一。然而,因為品種之間的遺傳差異較小,挖掘其多態性位點較為困難,特別是獲得穩定的序列特異性標記更為不易。利用簡單基因組測序可以獲得大量的差異位點。薄殼山核桃品種繁多,本研究中通過對大量基因組測序數據的分析,再加上PCR的反復驗證,獲得了一批可用于薄殼山核桃品種鑒定的SCAR 標記。雖然后續PCR 篩選的工作量較大,但SCAR 標記有其明顯的優點,比如可以通過普通的PCR 檢測條帶的有無,使用成本較低。研究結果也表明,400 多對引物擴增率雖然較高,但多態性比例并不高,因為這種簡單基因組測序是通過酶切后進行的,測序的信息主要來源于酶切位點附近,如果品種的差異點正好出現在酶切位點上,那么測序就會出現差異條帶,但事實上由此設計的引物能在所有品種中擴增出相似的條帶,這也是該方法的局限性造成的。同時,也可以利用簡單基因組測序獲得大量的SNP 位點,研究初期由于參考了核桃基因組數據,未能得到可靠的SNP 數據。因此,現有這些測序數據有進一步挖掘的潛力,比如利用薄殼山核桃品種87MX3-2.11 作為參考基因組[15],后續將進一步分析SNP 位點。
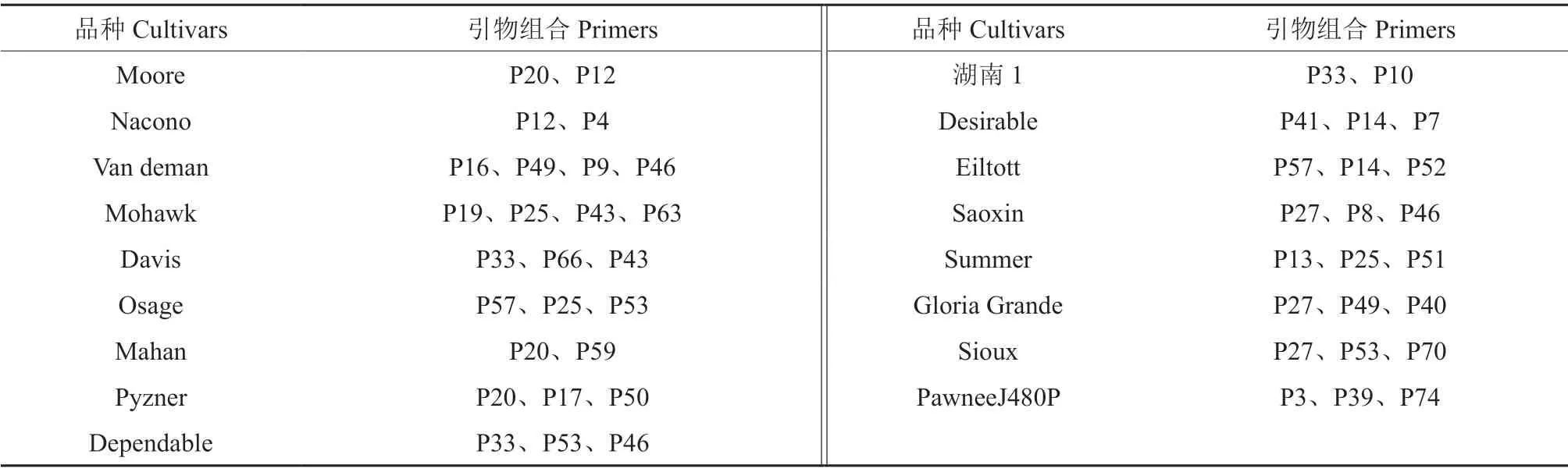
表4 用于薄殼山核桃品種鑒定的特異SCAR 標記組合舉例Table 4 The combination of specific SCAR markers for identification of pecan cultivars
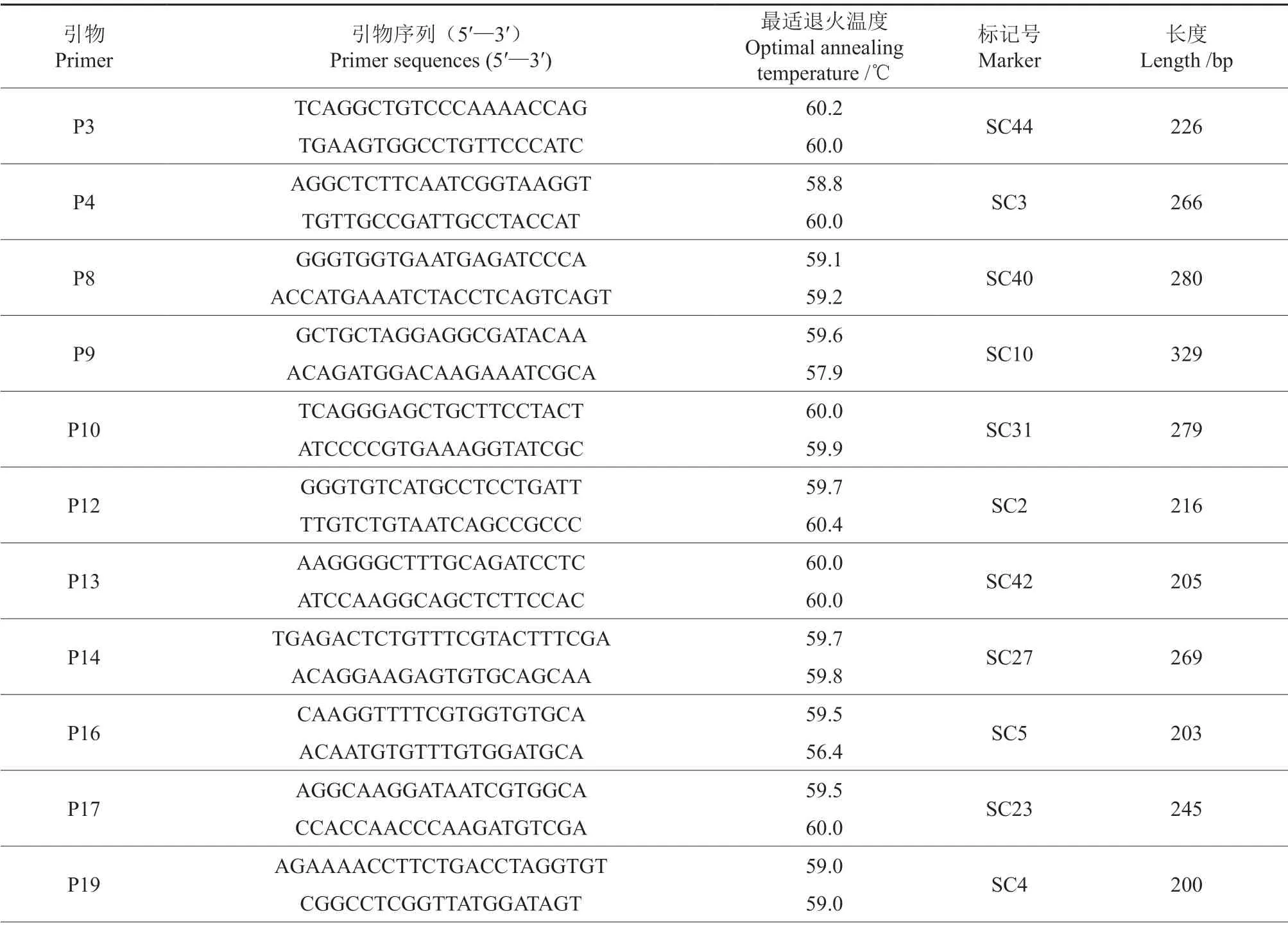
表5 用于薄殼山核桃品種鑒定的特異SCAR 標記引物的具體信息Table 5 Detailed information of primers with specific SCAR markers for identification of pecan cultivars

續表5Continuation of Table 5
但是,PCR 擴增有時會出現一定的假陽性,SCAR 引物也不例外,有時條帶較弱,經多次重復才能擴增。其關鍵是提取DNA 的質量以及DNA的分裝等問題。DNA 提取方式是影響DNA 質量的重要因素,SSR 標記的擴增也是如此[16]。DNA的分裝也會影響標記擴增的穩定性。通常情況下,較高濃度的DNA 須稀釋后用于后續的擴增,而稀釋后經常會面臨分裝的問題,由于DNA 是大分子,分裝會帶來分配不均的問題,有時會影響到試驗的重復性。而這一問題經常被研究人員忽視。通過定量PCR 試驗證實,如果未較好地進行分裝,那么即使是同一初始模板和同一引物分裝在不同PCR 管中也會有一定的擴增差異。因此在分裝DNA 樣品時,應進行必要的均質化,如超聲波震蕩等,對標記擴增的一致性有重要的作用。另外,擴增片段的大小也有重要影響,小片段更容易獲得具有一致性的擴增結果。就鑒定的方便程度來說,可以通過大小片段的組合進行混合引物PCR,但為了取得較為穩定的檢測結果,應測試各引物的不同濃度組合[17]。另外,在本研究中有些引物能產生多條特異性片段,其在品種鑒定中可能具有獨特的價值,但是鑒于其多態性較為復雜,本研究中暫未考慮,這些位點有待深入研究。
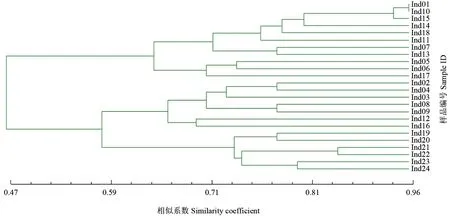
圖4 基于80 對引物的24 個薄殼山核桃樣品的聚類結果Fig.4 Cluster analysis of 24 C.illinoensis samples based on 80 pairs of primers
利用分子標記進行聚類分析,有助于分析品種間的親緣關系,對于后續的雜交親本選育具有重要意義。本研究中利用分離到的80 個SCAR 標記對現有24 種樣品進行分析,因為標記數量不夠多等原因,其結果存在與現有親緣關系不一致的問題,而來自薄殼山核桃品種園的雜交品種較多,后續將采集更多的品種進行類似的遺傳分析。
