淺低溫對心肌缺血-再灌注損傷合并膿毒癥大鼠模型的心肌保護作用
2021-05-02 01:57:08秦竹韻申世軒曲開勇曹芳芳張海濤
中國循環雜志 2021年4期
秦竹韻,申世軒,曲開勇,曹芳芳,張海濤
急性心肌梗死(AMI)與膿毒癥均是造成全球主要疾病負擔的原因[1-2]。AMI患者進行血管再通治療后合并膿毒癥在心臟重癥監護室常見。一項前瞻性研究表明,AMI合并膿毒癥的患者死亡風險較單純AMI患者顯著增加[3]。
AMI是由于冠狀動脈長時間閉塞所導致,而血流的恢復也會對心肌造成損傷。部分AMI患者因病情較重而導致住院時間延長,易發生院內感染和發展為膿毒癥。膿毒癥是宿主機體感染后所致的一系列綜合征,可導致包括心臟在內的多器官功能障礙[4]。心肌缺血-再灌注(I/R)損傷合并膿毒癥可造成心肌的氧化應激損傷、炎癥和凋亡。煙酰胺腺嘌呤二核苷酸磷酸氧化酶2(NOX2)是在心肌梗死以及膿毒癥中均可產生的一種主要的活性氧物質[5-6]。腫瘤壞死因子-α(TNF-α)可反映心肌I/R損傷合并膿毒癥中心肌炎癥情況[7]。此外,胱天蛋白酶-3(caspase-3)的表達增加可反映凋亡的發展[8]。因疾病機制復雜,心肌I/R損傷合并膿毒癥目前的臨床治療效果欠佳,目前尚少見有關其治療的文獻報道。
淺低溫(32℃~35℃)能降低機體代謝,促進損傷恢復。在動物模型中,淺低溫分別被證實可改善心肌I/R損傷和膿毒癥的癥狀[9-10]。因此,淺低溫對心肌I/R損傷合并膿毒癥可能也有保護作用。本研究通過建立SD大鼠的心肌I/R損傷合并膿毒癥疾病模型,從心功能、心肌梗死面積、心肌損傷、氧化應激、炎癥和凋亡方面探究淺低溫對心肌的保護作用。
1 材料與方法
1.1 實驗動物及分組
選用健康雄性SD大鼠15只,體重400~600 g,所有動物購于北京維通利華實驗動物技術有限公司。實驗前予以大鼠清潔飲食,觀察7 d,無異常反應。大鼠隨機分為3組:心肌I/R損傷合并膿毒癥淺低溫(MHT)組,心肌I/R損傷合并膿毒癥常溫(NT)組,假手術常溫組作為對照組,每組大鼠選用5只。
1.2 心肌I/R損傷合并膿毒癥模型制作
心肌I/R損傷模型按常規方法建立[11]。首先結扎前降支約30 min后抽出尼龍線使血管再通建立缺血再灌注損傷模型,然后抽出尼龍線同時將內毒素(LPS,O111B4,Sigma公司,美國)15 mg/kg腹腔注射至大鼠體內建立膿毒癥模型,對照組注射等量生理鹽水(NS)。大鼠基礎左心室收縮壓95~115 mmHg(1 mmHg=0.133 kPa),約15 min后下降至基礎值60%~70%左右(約60~80 mmHg)并維持1 h以上表示膿毒癥模型建立成功[12]。
1.3 溫度控制
實驗中大鼠的正常體溫在(37.0±0.5)℃,核心體溫通過檢測肛溫實現。注入LPS或生理鹽水同時,將MHT組大鼠置于冰床上降溫,每隔5 min檢測一次肛溫,15 min降至目標溫度(33.0±0.5)℃,然后維持2 h。NT組及對照組實驗全程用加熱手術臺及紅外線燈照射維持在常溫,防止大鼠因麻醉所導致的體溫丟失。
1.4 觀察指標
目標溫度維持2 h后,抽取靜脈血并處死大鼠取心臟。檢測大鼠心肌損傷標志物、心肌病理變化、心肌梗死面積、氧化應激、凋亡和炎癥的情況。
1.4.1血流動力學指標 :實驗全程用Powerlab系統記錄心率、左心室最大收縮壓、左心室舒張末期壓力(LVEDp)、收縮期壓力最大變化速率(+max dp/dt)及舒張期壓力最小變化速率(-min dp/dt)等血流動力學指標的變化。
1.4.2心肌損傷標志物:經頸靜脈抽血用酶聯免疫吸附測定法檢測乳酸脫氫酶(LDH)、肌酸激酶同工酶(CK-MB)含量。將血樣離心取上清,加樣,封板后置 37℃孵育 60 min,再洗滌、拍干,然后加入顯色劑于37℃暗處反應 20 min,終止反應后測定吸光度。
1.4.3心肌病理學變化:將大鼠結扎部位以下心肌組織放置在4%多聚甲醛溶液中固定。經乙醇梯度脫水,石蠟包埋,切片,進行蘇木精-伊紅(HE)染色,然后用光學顯微鏡觀察心肌組織病理形態學變化。
1.4.4心肌梗死面積測定:目標溫度維持2 h后,將1 ml 1%氯化三苯基四氮唑(TTC)及2 ml 1%伊文思藍注入大鼠頸靜脈將心臟染色,然后取材。粉紅色區域為梗死區,藍色為非缺血心肌區域。將心臟快速凍至-80℃ 20 min后取出,切成5個約2 mm薄片。圖片用Image Pro Plus 6.0軟件測量心肌梗死范圍,心肌梗死程度用梗死面積占總面積比例表示。
1.4.5免疫印跡法檢測NOX2表達:取 20 mg大鼠心臟組織加入裂解液提取總蛋白,BCA試劑盒進行蛋白定量。聚丙烯酰胺凝膠電泳后,轉至聚偏氟乙烯膜,用5%脫脂奶的 TBST液封閉 1 h,洗滌 3次,然后用兔抗鼠 NOX2抗體及兔抗鼠 β-肌動蛋白(β-actin)單克隆抗體(1∶2 000稀釋)孵育,4℃過夜,加入辣根過氧化物酶標記的羊抗兔 IgG(1∶2 000稀釋),化學發光法進行成像。β-actin作為內參蛋白。用 Image J軟件分析條帶灰度值,計算NOX2和β-actin比值作為NOX2的相對表達量。
1.4.6免疫組化檢測caspase-3和TNF-α 表達:基于HE染色結果,包含心肌梗死區域的心肌組織用石蠟包埋、封閉后,用抗TNF-α 和caspase-3抗體孵育于4℃,過夜。在室溫下用二抗孵育1 h。將3,3’-二氨基聯苯胺/過氧化氫加到切片表面,室溫染色10 min,然后成像。計算caspase-3或TNF-α 陽性細胞背景與總細胞背景的比值作為caspase-3或TNF-α 相對表達量。
1.5 統計學方法
實驗結果采用SPSS 26.0軟件進行處理,計量資料用均數±標準差()表示。兩組間均數比較使用獨立樣本t檢驗,多組間比較使用重復測量的方差分析,兩兩比較用LSD法(方差齊時)或用Dunnett T3法(方差不齊時),非正態分布資料比較用 Kruska-Wallis檢驗。P<0.05為差異有統計學意義。
2 結果
2.1 淺低溫對血流動力學的影響(表1)
MHT組再灌注2 h心率較NT組降低[(396.63±39.93)次/min vs.(457.74±32.77)次/min,P<0.05];左心室最大收縮壓升高[(129.14±34.05)mmHg vs.(85.63±14.74)mmHg,P<0.05],差異均有統計學意義;MHT組+max dp/dt再灌注2 h較NT組及對照組升高,但差異無統計學意義;LVEDp和-min dp/dt再灌注2 h絕對值也較NT組及對照組均有所增加,但差異均無統計學意義(P均>0.05)。
2.2 淺低溫對心肌損傷的影響(表2、圖1)
NT組LDH及CK-MB顯著高于對照組,差異均有統計學意義(P均<0.05)。MHT組LDH及CK-MB均高于對照組,但差異均無統計學意義(P均>0.05)。MHT組LDH及CK-MB均顯著低于NT組,差異均有統計學意義(P均<0.05)。HE染色光鏡下MHT組心肌細胞相對完整,心肌壞死呈點狀或灶狀,心肌間質淤血程度輕且炎細胞浸潤較少。NT組的心肌出現水腫、灶狀或片狀壞死,心肌間質區淤血程度重及存在大量炎細胞浸潤。對照組心肌細胞排列整齊,結構完整。
2.3 淺低溫對心肌梗死面積的影響
對照組心肌無明顯缺血區,MHT組及NT組出現心肌梗死。MHT組與NT組相比,心肌梗死面積比率明顯減少[(11.23±2.82)% vs.(19.25±4.45)%,P<0.05],差異有統計學意義。
2.4 淺低溫對心肌氧化應激的影響(表3、圖2)
免疫印跡法檢查結果顯示,NT組及MHT組NOX2表達少量增加。NT組NOX2的相對表達量較對照組增加,差異有統計學意義(P<0.05)。MHT組NOX2的相對表達量較對照組增加,差異無統計學意義(P>0.05)。MHT組較NT組相對表達量減少,但差異亦無統計學意義(P>0.05)。
表1 各組大鼠不同時間段的血流動力學數據變化(n=5,)
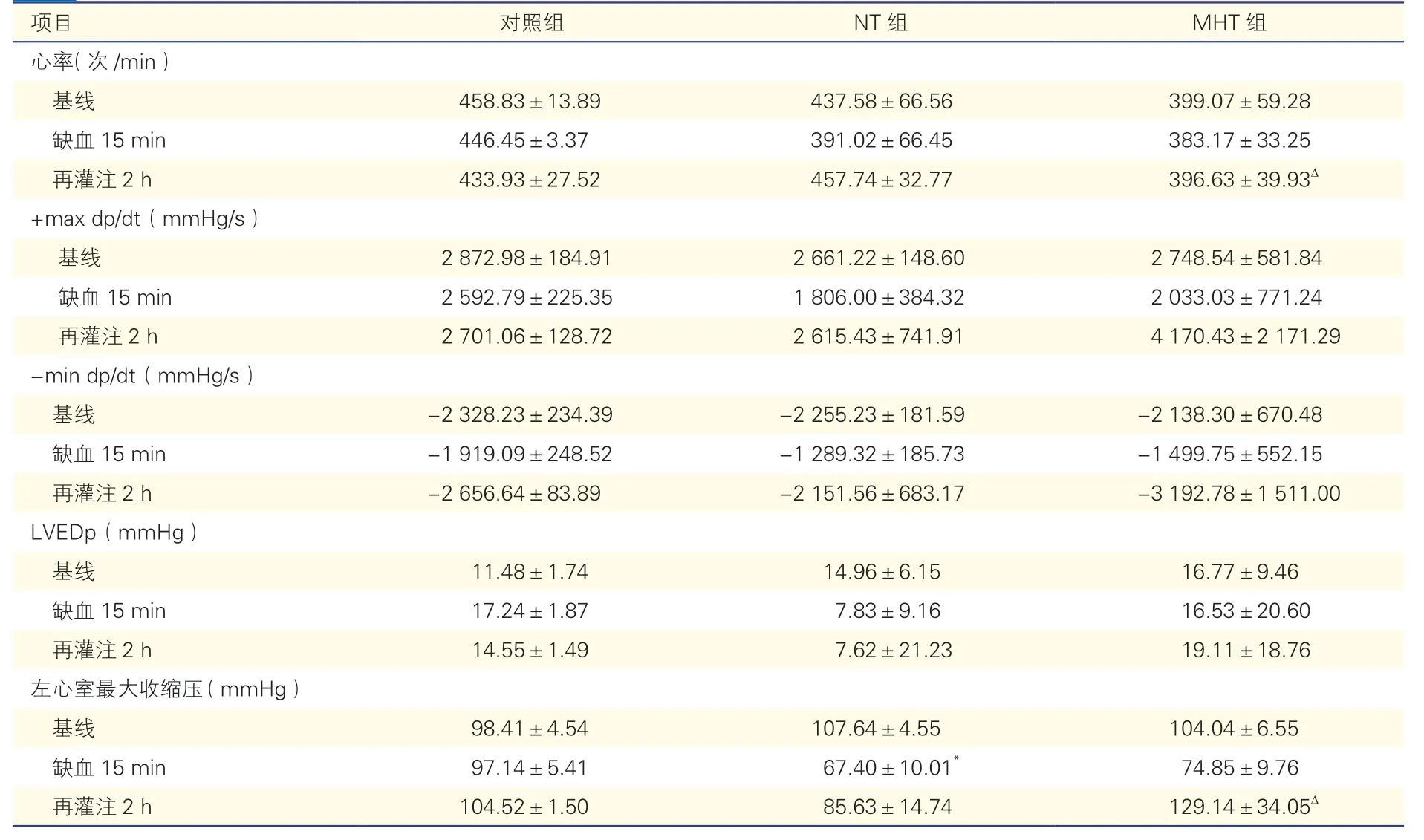
表1 各組大鼠不同時間段的血流動力學數據變化(n=5,)
注:NT:常溫;MHT:淺低溫;+max dp/dt:收縮期壓力最大變化速率;-min dp/dt:舒張期壓力最小變化速率;LVEDp:左心室舒張末期壓力。與對照組比較*P<0.05;與NT組比較Δ P<0.05。1 mmHg=0.133 kPa
表2 淺低溫維持2 h后各組大鼠LDH和CK-MB水平比較(U/L,n=5,)

表2 淺低溫維持2 h后各組大鼠LDH和CK-MB水平比較(U/L,n=5,)
注:NT:常溫;MHT:淺低溫;LDH:乳酸脫氫酶;CK-MB:肌酸激酶同工酶。與對照組比較*P<0.05;與NT組比較△P<0.05
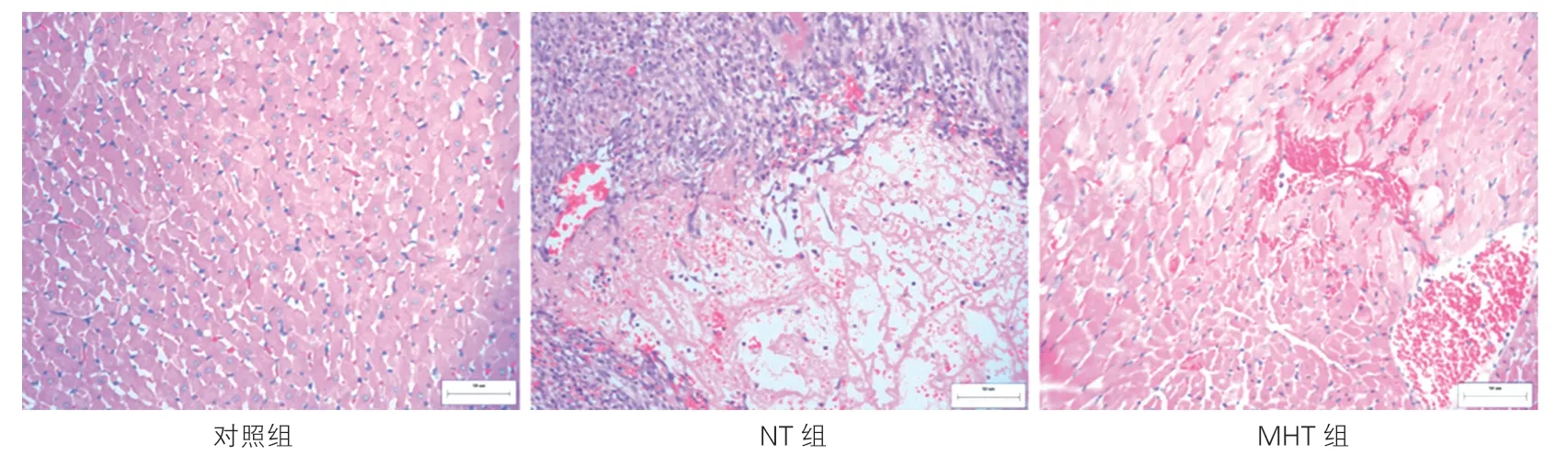
圖1 淺低溫維持2 h后光鏡下各組大鼠心肌組織病理學變化(×200)
表3 淺低溫維持2 h后各組大鼠NOX2、caspase-3及TNF-a相對表達量(%,n=5,)

表3 淺低溫維持2 h后各組大鼠NOX2、caspase-3及TNF-a相對表達量(%,n=5,)
注:NT:常溫;MHT:淺低溫;NOX2:煙酰胺腺嘌呤二核苷酸磷酸氧化酶2;caspase-3:胱天蛋白酶-3;TNF-α:腫瘤壞死因子-α。與對照組比較*P<0.05;與NT組比較△P<0.05
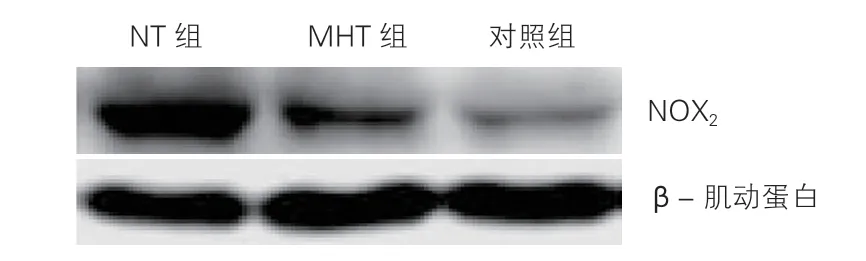
圖2 淺低溫維持2 h后各組大鼠免疫印跡法NOX2表達情況
2.5 淺低溫對心肌凋亡及炎癥的影響(表3、圖3)
免疫組化檢測NT組caspase-3及TNF-a的相對表達量較對照組顯著增高,差異均有統計學意義(P均<0.05)。MHT組caspase-3及TNF-a的相對表達量均較對照組升高,但差異均無統計學意義(P均>0.05)。MHT組caspase-3及TNF-a的相對表達量較NT組降低,差異均有統計學意義(P均<0.05)。
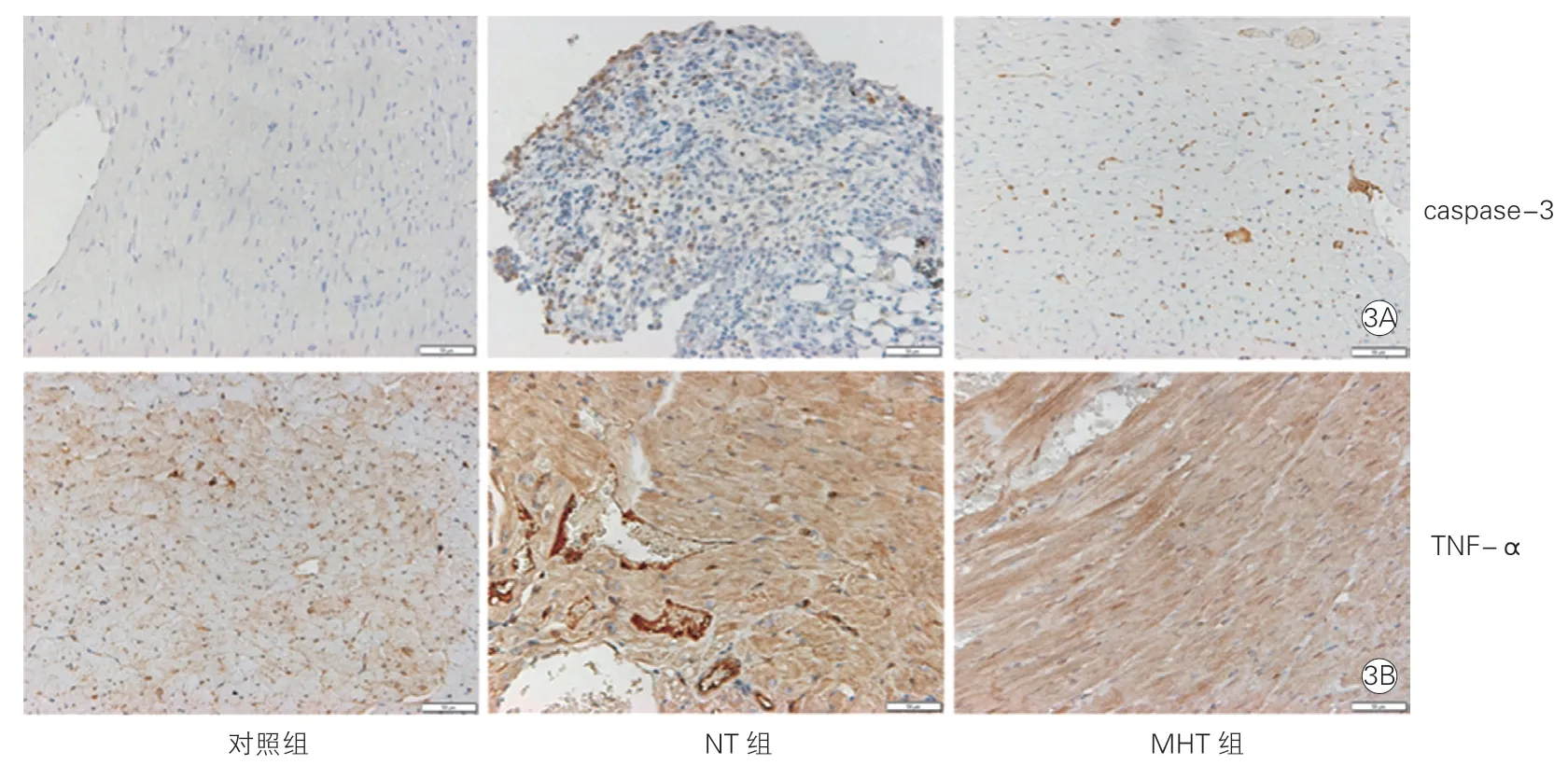
圖3 淺低溫維持2 h后光鏡下各組大鼠免疫組化分析中caspase-3(3A)及TNF-α(3B)的表達(×200)
3 討論
本研究成功建立SD大鼠心肌I/R損傷合并膿毒癥模型,并通過在模型中應用淺低溫證實其可改善心肌的收縮和舒張功能,減少心肌梗死面積,減輕心肌損傷,也有抗心肌氧化應激、凋亡和炎癥反應的作用。
淺低溫在其他動物實驗中已被證實有正性肌力的作用,可以改善心臟收縮及舒張功能。Huang等[13]在一項將心跳驟停的動物實驗中應用淺低溫,發現+max dp/dt、-min dp/dt和心輸出量增加,提示心臟收縮及舒張功能均有改善,本研究結果與此類似。本研究中的MHT組中,心率較前降低,但LVEDp、左心室最大收縮壓、+max dp/dt及-min dp/dt均有上升,提示大鼠的心輸出量增加。而NT組中雖然心率增加,但心臟的舒縮壓及速率均較基線減少,提示大鼠心功能受損。這證實在心肌I/R損傷合并膿毒癥中存在心功能損害,淺低溫有改善心臟收縮及舒張功能的作用。然而,MHT組及淺低溫維持2 h后+max dp/dt、LVEDp和-min dp/dt結果較NT組及對照組差異均無統計學意義,究其原因可能是因為大鼠樣本較少,或低溫時間需延長等。
既往動物實驗表明,淺低溫可減少心肌I/R損傷的梗死面積,但對于降溫時機存在爭議。Dash等[14]的一項在豬心肌I/R損傷模型研究中,降溫時機選在再灌注前30 min,結果示心肌梗死面積明顯減少。Kanemoto等[15]研究顯示,在開始再灌注時降溫,可最大限度減少心肌梗死面積。Marek-Iannucci等[16]在心肌I/R損傷豬模型中選擇再灌注后30 min降溫,心肌梗死面積也有所減少。然而,Hale等[17]在兔心肌I/R損傷模型中將降溫時機應用在再灌注后30 min,結果顯示雖然淺低溫可改善無復流現象,但心肌梗死面積并未減少。在本研究中,降溫時機在再灌注同時,MHT組的心肌梗死面積較NT組心肌梗死面積顯著減少,這與Kanemoto等[15]研究結果一致,提示即使在心肌I/R損傷合并膿毒癥的情況下,在再灌注治療同時降溫也可減少心肌梗死面積。
本研究顯示,淺低溫減少心肌梗死面積是通過減輕心肌損傷實現的。心肌損傷時,包括LDH和CK-MB在內的心肌損傷標志物釋放入循環中,LDH和CK-MB的升高可協助AMI的診斷,其升高程度可代表心肌受損程度[18]。本實驗中,MHT組中的LDH和CK-MB較NT組減低,且MHT組和對照組升高程度相比差異無統計學意義,證明淺低溫可以緩解心肌損傷。此外,在病理學層面上,HE染色直接顯示MHT組心肌受損程度較NT組明顯減輕,心肌壞死較NT組少,且炎細胞浸潤較少。
NOX2是AMI和膿毒癥中均可產生的氧化應激相關物質[5-6]。本研究顯示NOX2在MHT組較NT組表達減少,但差異無統計學意義,可能與樣本量較少有關。此外,在本實驗中,心肌I/R損傷合并膿毒癥導致提示炎癥反應的炎癥標志物TNF-α 及提示心肌細胞凋亡的caspase-3升高,而淺低溫有改善心肌炎癥及抗心肌細胞凋亡的作用。
綜上所述,本研究從心功能、心肌梗死面積、心肌損傷程度、氧化應激、炎癥及凋亡幾方面證實了淺低溫在心肌I/R損傷合并膿毒癥的心肌保護作用,為臨床心肌I/R損傷合并膿毒癥的危重患者的治療提供了動物研究基礎。然而,本實驗通過注射LPS建立的膿毒癥模型與臨床上病原微生物導致的膿毒癥有區別,且大鼠與人之間物種之間的差別可導致結果不一致,故淺低溫對為臨床心肌I/R損傷合并膿毒癥患者的有效性有待進一步研究。
利益沖突:所有作者均聲明不存在利益沖突
猜你喜歡
童話王國·奇妙邏輯推理(2024年5期)2024-06-19 16:03:38
新少年(2022年9期)2022-09-17 07:10:54
音樂探索(2022年2期)2022-05-30 21:01:37
小天使·一年級語數英綜合(2020年6期)2020-12-16 02:56:41
中學生數理化·七年級數學人教版(2020年10期)2020-11-26 08:24:50
數學物理學報(2020年2期)2020-06-02 11:29:24
小天使·一年級語數英綜合(2019年8期)2019-08-27 02:23:00
小學科學(學生版)(2018年7期)2018-08-13 09:33:04
光學精密工程(2016年6期)2016-11-07 09:07:19
北極光(2014年8期)2015-03-30 02:50:51
