毛囊角化病九例ATP2A2基因突變研究
2021-05-11 11:15:48于學萍夏倩倩孫樂樂糜自豪王真真龐于功奇付希安張福仁
中國麻風皮膚病雜志 2021年5期
關鍵詞:數據庫
于學萍 趙 晴 夏倩倩 孫樂樂 糜自豪 王真真龐 政 于功奇 付希安 劉 紅 張福仁,
1濱州醫學院,山東煙臺,264033;2山東第一醫科大學附屬皮膚病醫院(山東省皮膚病醫院,山東省皮膚病性病防治研究所),濟南,250022
毛囊角化病(DD)是一種常染色體顯性遺傳性皮膚病,盡管其表型是可變的,但是具有較高的外顯率[1]。DD的臨床特征為脂溢性區域的疣狀丘疹和斑塊、有掌跖角化性凹點和甲營養不良[2]。1993年Craddock等[3]將DD的致病基因突變定位到染色體12q23-q24.1,1999年Sakuntabhai等[4]發現編碼肌漿/內質網鈣離子-ATP酶2(SERCA2)的ATP2A2基因與DD之間存在很強的相關性。SERCA2是內質網應激的啟動因子,在Ca2+穩態中發揮重要作用[5]。到目前為止,已經報道了ATP2A2基因上超過298個變異引起的DD,但沒有發現熱點突變區域(http://www.lovd.nl/ATP2A2)。根據以前的文獻,通過Sanger測序檢測到60%~88% DD患者在ATP2A2上有突變[5]。然而,其余12%~40%患者的病因尚未闡明。我們在臨床上收集了9例DD患者,通過Sanger直接測序法和全外顯子組測序(WES)對患者進行突變檢測,確定了基因突變位點。
1 對象與方法
1.1 研究對象 本研究選取來自我院皮膚科門診的9例患者,最小發病年齡14歲,最大91歲,患者經臨床及病理組織檢查,確診為DD,部分患者皮損見圖1。患者簽署知情同意書并收集病史資料及外周血。同時選取100名健康人作為對照。
1.2 研究方法
1.2.1 DNA提取 本研究經山東省皮膚病性病防治研究所倫理委員會批準,簽署知情同意書后,抽取患者及其家屬外周血5 mL,同時采集100名無親緣關系的正常個體外周血5 mL作為對照,-80℃保存。采用DNA提取試劑盒(天根生化科技有限公司,北京)提取基因組DNA,并用全波長紫外/可見光掃描分光光度計ND-8000(Thermo Fisher公司,美國)測定其純度及含量,將其標化為30 ng/μL的DNA模板。
1.2.2 Sanger測序 在NCBI網站(https://www.ncbi.nlm.nih.gov/tools/primer-blast/)上設計了19對引物,通過聚合酶鏈式反應(PCR)擴增ATP2A2基因全部21對外顯子及側翼序列,引物交由北京華大基因科技有限公司合成。25 μL PCR反應體系為:PCR taq mix 12.5 μL,dNTP上下游引物各1 μL,DNA模板(30 ng/μL)0.5 μL,ddH2O 10 μL。PCR反應條件:預變性94℃ 10 min,變性94℃ 30 s,退火55~59℃ 45 s,延伸72℃ 10 min,共進行35個循環,最后72℃延伸1 min。PCR產物經1.2%瓊脂糖凝膠電泳檢測。DNA 測序PCR產物經醋酸鈉-乙醇沉淀法純化后直接于ABI 3130XL基因分析儀(Applied Bio-systems,美國)上進行測序。突變分析所有測序結果經NCBI blast(http://blast.ncbi.nlm.nih.gov/Blast.cgi)數據庫進行分析比對,用SIFT及Polyphen-2軟件對基因突變進行功能預測。
1.2.3 全外顯子組測序 對于不攜帶ATP2A2突變的患者,利用Biorupter(Diagenode,比利時)剪切每個樣本的基因組DNA(200 ng),得到150~200 bp的片段。使用AIExome富集試劑盒V1(iGeneTech,中國北京)進行全外顯子組測序,使用Illumina HiSeq 6000平臺對150 bp配對端進行大規模平行測序。使用BWA v0.7.17程序將得到的reads映射到hg19/GRCh37人類參考基因組。隨后,按照最佳實踐執行了GATK v4.0.12.0來調用變體。采用bcftools對不同過濾步驟進行篩選和過濾。
1.2.4 突變分析 在兩個公共變異數據庫(1000 Genomes Project和Exome Aggregation Consortium)中過濾突變,以去除1000 Genomes Project中存在的突變或Exome Aggregation Consortium數據庫中最小等位基因頻率(MAF)大于0.01%的突變。對于篩選出來的變異,當錯義突變在四種預測算法(SIFT、polyphen2、LRT和MutationTaster)中的至少三種中被預測為有害時,則被定義為具有有害突變。其他突變類型,如剪接位點突變、移碼刪除/插入和非移碼刪除/插入都被認為是破壞性的。最后,過濾后的突變在364個內部對照中得到進一步驗證。
2 結果
通過對9例患者ATP2A2基因的直接測序,我們在5例DD患者中發現了1例已報道的突變和3例新突變(圖2),包括2例非移碼刪除/插入(c.24_26delGGT,c.823_824insTAA)、1例剪接位點突變(c.119-2A>G)和1例錯義突變(c.2803T>C)(表1)。在100個健康對照和兩個公共變異數據庫(1000 genome Project和Exome Aggregation Consortium)中均未檢測到這些突變。
對ATP2A2無突變的患者進行WES檢測,共發現了4個ATPase基因的4個突變,包括ITPR3基因的1個剪接位點突變(c.4789-3C>G),PLCB1(c.1666G>A)、RYR1(c.2179C>T)和ATP2B2(c.707T>C)基因上3個錯義突變,通過公共數據庫(1000 genome Project和Exome Aggregation Consortium)進行篩選后,ITPR3、PLCB1、RYR1上的3個突變均未見報道。此外,這些變體在364個內部對照中沒有發現。ATP2B2基因上的突變(c.707T>C)在Exome Aggregation Consortium數據庫中被檢測到,而在其他兩個數據庫中則不存在(1/4326)(表2)。通過Sanger測序進一步證實了這些突變的存在(圖3)。
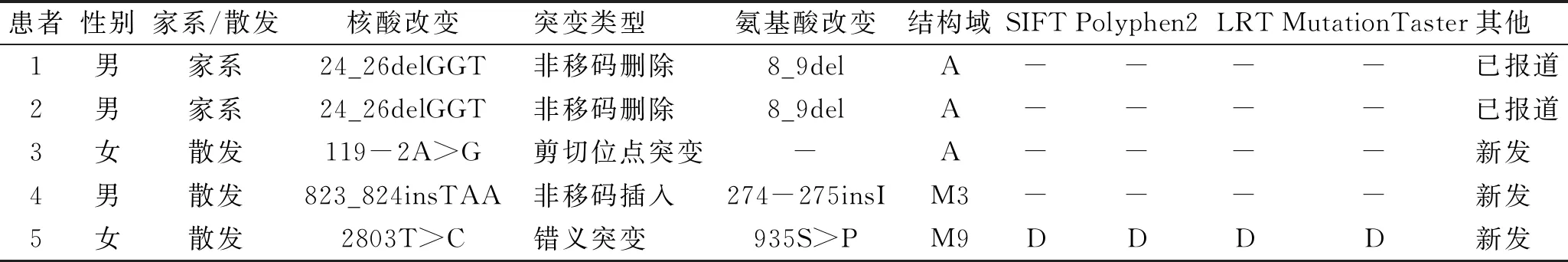
表1 5例DD患者中發現的可能致病的ATP2A2變異
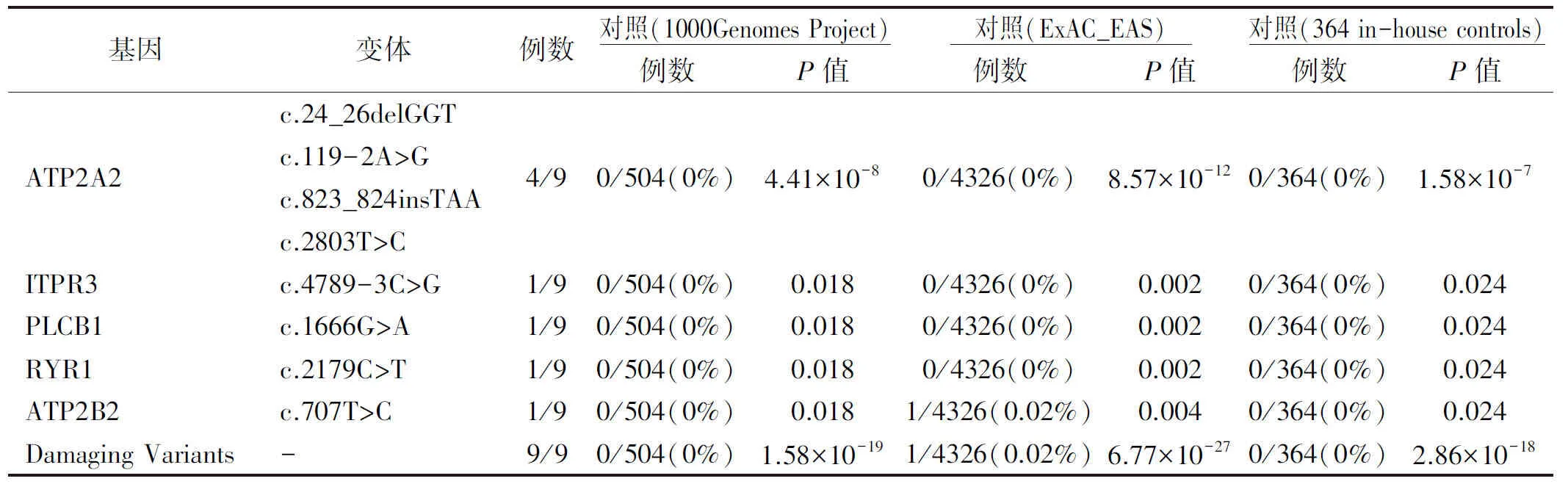
表2 突變頻率分布

圖1 1a、1b:頸部、腋窩部可見實質性小丘疹;1c:胸腹部散發褐色角化性丘疹,上覆褐色油膩性痂皮,部分融合成疣狀斑塊

2a:1號外顯子突變 c.24_26delGGT; 2b:剪切位點突變 c.119-2A>G; 2c:8號外顯子突變 c.823_824insTAA; 2d:19號外顯子突變2803T>C

圖3 4例DD患者其他基因中SNP的鑒定(c.4789-3C>G, c.1666G>A, c.2179C>T, c.707T>C)
3 討論
DD是一種常染色體顯性遺傳病,由表皮鈣穩態異常引起。1999年Sakuntabhai等[4]發現ATP2A2基因與DD之間存在很強的相關性。人類ATP2A2基因編碼SERCA2蛋白,SERCA2將鈣離子從胞漿轉移至內質網內并對表皮的細胞連接和分化起重要作用[6]。ATP2A2基因長約76 Kb,包含 21 個外顯子,由5個主要結構域組成,包括一個跨膜結構域(M),由11個跨膜蛋白(M1-M11)組成,5個連接區域(S1-S5)和3個細胞質結構域,其包括一個執行元件域(A)、一個核苷酸ATP結合結構域(N)和一個磷酸化結構域(P)[7]。自1999年以來,ATP2A2至少有298個突變被報道。然而,所有的變體在ATP2A2中分散分布,沒有特定的突變熱點[8],我們的發現與之前的研究一致。本文總結了報道的298個ATP2A2基因變異的分布。
在這些突變中,約5%(9/298)的突變破壞了M1和M2結構域,這是細胞質中高度保守的TGE基序,在ATP水解向Ca2+轉運的能量傳遞過程中起至關重要的作用[4]。我們報道的ATP2A2 M3結構域的非移碼插入c.823_824insTAA影響了管腔鈣通道的運輸功能[9]。約18.8%(56/298)的突變位于M4、M5、M6和M8,這些區域包含Ca2+結合位點,這些突變可導致Ca2+轉運功能的完全喪失[10]。在M9和M10區域發現了大約3.3%(10/298)的突變,包括我們發現的突變(c.2803T>C),這可能影響ER膜的錨定[9]。報道的A、P和N域變異分別占21.5%(64/298)、15.1%(45/298)和20.8%(62/298)。在本研究中,兩個新突變c.24_26delGGT和c.119-2A>G位于A結構域,在鈣離子結合和改變位置方面發揮了重要作用[11]。此外,有28個突變(9.4%)位于細胞質連接區(S1-S5),這些區域輔助離子的轉運[12]。
SERCA2可以調節Ca2+穩態。Ca2+信號通路在ER的鈣隔離、鈣黏蛋白的定位中起著至關重要的作用,因此Ca2+信號通路的失調可能導致DD的發生[13]。此外,SERCA2作為一種ATP酶,也通過Ca2+信號通路調節細胞間的粘附和鈣穩態[14]。在本研究中,我們在4例無ATP2A2突變的DD患者中發現了4個ATP酶基因上的有害突變,且均參與了Ca2+信號通路的。Ca2+信號的產生包括1,4,5-三磷酸受體(Ins(1,4,5)P3R)和ryanodine受體(RyR)家族Ca2+通道的打開,隨后Ca2+從內質網中迅速釋放[15]。細胞外增加的Ca2+結合位于質膜的Ca2+受體(CaR),產生第二信使肌醇-1,4,5-三磷酸(IP3),通過與ITPR3結合導致內質網Ca2+釋放[8]。同時,由ATP2B2編碼的PMCA2蛋白和SERCA2蛋白均屬于p型Ca2+蛋白ATP酶家族,被認為是高度保守的磷酸化DKTGT序列,該酶還參與Ca2+振蕩的產生和細胞質Ca2+濃度的恢復[7]。因此,基于我們的研究結果,我們認為參與Ca2+信號通路的ATP酶基因的突變可能導致內質網和高爾基體Ca2+釋放不足,從而導致鈣紊亂并引起臨床癥狀。然而,潛在的機制還沒有被證明,我們需要增加樣本數量,進一步進行功能測試來驗證。
本文通過Sanger測序在5例DD患者中鑒定了3個新的ATP2A2基因變異和1個之前報道的突變。此外,在4例ATP2A2無突變的患者中,我們通過WES分析發現了分別位于4個ATP酶基因(ITPR3、PLCB1、RYR1、ATP2B2)的4個突變,這可能是導致本病的原因。我們的發現擴大了ATP2A2突變的范圍,并為毛囊角化病提供了新的可能的發病機制。
猜你喜歡
財經(2017年15期)2017-07-03 22:40:49
財經(2017年2期)2017-03-10 14:35:35
華東師范大學學報(自然科學版)(2017年1期)2017-02-27 13:41:08
財經(2016年15期)2016-06-03 07:38:02
財經(2016年3期)2016-03-07 07:44:46
財經(2016年6期)2016-02-24 07:41:51
財經(2015年3期)2015-06-09 17:41:31
財經(2014年21期)2014-08-18 01:50:18
財經(2014年6期)2014-03-12 08:28:19
財經(2013年6期)2013-04-29 17:59:30
