基于苯并三唑主鏈的一系列N-型共軛聚電解質(zhì)的合成方法
2021-05-18 02:12:06
科教導(dǎo)刊·電子版 2021年9期
(杭州電子科技大學(xué)材料科學(xué)與工程學(xué)院 浙江·杭州 310018)
1 背景研究
共軛聚電解質(zhì)的離子側(cè)鏈可以提供離子遷移、偶極效應(yīng)和溶于極性溶劑(如水,甲醇等)和實現(xiàn)多層電子器件制備,因此共軛聚電解質(zhì)廣泛應(yīng)用于各類有機(jī)光伏器件中,如有機(jī)聚合物太陽能電池、場效應(yīng)晶體管、發(fā)光二極管及熱電應(yīng)用。另外,離子側(cè)鏈還能被作為誘導(dǎo)共軛聚電解質(zhì)聚集的設(shè)計手段。
影響共軛聚電解質(zhì)光電性能的因素很多,包括共軛聚合物主鏈因素。含苝二酰亞胺、二酮吡咯并吡咯、苯并噻二唑和苯并三唑等缺電子雜環(huán)主鏈結(jié)構(gòu)單元的聚合物在有機(jī)電子設(shè)備中能提供n-型的電荷傳輸行為。然而該類含電子缺失共軛單元主鏈的共軛聚電解質(zhì)的合成較難。相比之下,苯并噻二唑和苯并三唑等缺電子雜環(huán)主鏈結(jié)構(gòu)單元的共軛聚電解質(zhì)的合成可行性較為樂觀。以苯并噻二唑和苯并三唑主鏈結(jié)構(gòu)單元的共軛聚電解質(zhì)由于它們較強(qiáng)的電子親和力而備受關(guān)注,然而有文獻(xiàn)報道表明基于苯并噻二唑和苯并三唑共軛主鏈的聚合物的溶解性極差,且其分子量很低(一般<5000g/mol)。另外,以苯并噻二唑和苯并三唑結(jié)構(gòu)單元主鏈的共軛聚電解質(zhì)的光電性能單從其分子結(jié)構(gòu)很難預(yù)測,因為它還依賴于其溶液狀態(tài)的分子聚集態(tài)。
共軛聚電解質(zhì)的一般合成方法是將非離子共軛聚合物的側(cè)鏈官能團(tuán)(即鹵素)轉(zhuǎn)化為離子官能團(tuán)。目前共軛聚電解質(zhì)合成常用的方法是Suzuki偶聯(lián)聚合反應(yīng)。然而這種合成方法在制備含缺電子結(jié)構(gòu)單元的聚合物方面效率較低。此外,我們還發(fā)現(xiàn)基于苯并噻二唑和苯并三唑共軛主鏈的前體聚合物上官能團(tuán)的定量轉(zhuǎn)化是具有挑戰(zhàn)性的。
本文的一個目的是提供一種基于苯并三唑主鏈的一系列N-型共軛聚電解質(zhì)的合成方法。由于基于苯并噻二唑和苯并三唑缺電子主鏈結(jié)構(gòu)的非離子共軛聚合物的側(cè)鏈官能團(tuán)(即鹵素)不能完全轉(zhuǎn)化為離子官能團(tuán)。因此,我們發(fā)現(xiàn)一種一步合成共軛聚合物的方法,即從含離子側(cè)鏈的單體直接聚合成共軛聚電解質(zhì)。
本文的一個目的是提供一種合成以苯并三唑為主鏈的一系列N-型共軛聚電解質(zhì)的合成方法,該類共軛聚電解質(zhì)的合成只能從含離子側(cè)鏈的單體直接聚合成共軛聚電解質(zhì)。
2 合成的具體步驟
(1)使用Suzuki偶聯(lián)聚合反應(yīng)方法合成陽離子共軛聚電解質(zhì):PBTBTz-TMABr和 PBTBTz-PyrBr。
合成過程如圖1,共軛聚電解質(zhì)的合成過程是:手套箱的氮?dú)夥諊拢谖⒉ü苤蟹謩e加入組分2(1.0摩爾比)、組分1(1.0摩爾比)、催化劑四三苯基膦鈀(2%摩爾比)、四氫呋喃溶劑和一顆攪拌子,然后把微波管密封取出手套箱。把經(jīng)脫氣處理過的碳酸鈉水溶液注入微波管中。溶液在80℃條件下攪拌24小時后倒入甲醇中沉淀,把紅色沉淀物過濾,經(jīng)適量丙酮、甲醇溶劑洗滌后收集沉淀物。所收集的固體經(jīng)索氏提取器索氏提取,依次經(jīng)丙酮、正己烷洗滌,再經(jīng)氯仿洗滌收取干燥得中性聚合物PBTBTz-Br。經(jīng)GPC測試發(fā)現(xiàn)該方法所得到的PBTBTz-Br分子量為1196 g/mol(測試溶劑為氯仿)。把聚合物PBTBTz-Br分別置于三甲胺和吡啶溶液中攪拌進(jìn)行末端溴功能化處理,經(jīng)真空旋轉(zhuǎn)干燥得 PBTBTz-TMABr和PBTBTz-PyrBr。然而,所得到的共軛聚電解質(zhì) PBTBTz-TMABr和PBTBTz-PyrBr分子量雖低,但其溶解性很差,幾乎不溶于任何溶劑中。證明了基于苯并噻二唑和苯并三唑缺電子主鏈結(jié)構(gòu)的共軛聚電解質(zhì)合成方法從非離子共軛聚合物的側(cè)鏈官能團(tuán)(即鹵素)轉(zhuǎn)化為離子官能團(tuán)的過程是不可行的。
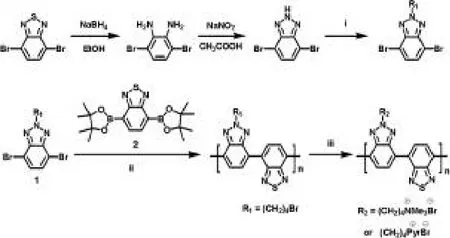
圖1:PBTBTz-TMABr和PBTBTz-PyrBr的合成過程。反應(yīng)過程:(i)4,7-二溴-2H-苯并三唑,1,4-二溴丁和氫氧化鉀在60℃條件下反應(yīng)5 h。(ii)四(三苯基膦)鈀,碳酸鈉,4,7-二溴-2(-6-溴-己基)-2H-苯并三唑和4,7-雙(-4,4,5,5-四甲基-[1,3,2]二氧雜環(huán)戊硼烷-2-基)-苯并[1,2,5]噻二唑在THF/H2O溶劑中、90℃條件下反應(yīng)24 h。(iii)末端含溴的側(cè)鏈共聚物置于THF/H2O溶劑中,加入三甲胺或者吡啶,在60℃條件下反應(yīng)24小時。
(2)使用Suzuki偶聯(lián)聚合方法合成PBTBTz-SO3Na。合成過程如圖2,共軛聚電解質(zhì)的合成過程是:手套箱的氮?dú)夥諊拢谖⒉ü苤蟹謩e加入組分3(1.0摩爾比)、組分4(1.0摩爾比)、碳酸鈉(5.0摩爾比)和催化劑四三苯基膦鈀(2%摩爾比)和一顆攪拌子,然后把微波管密封取出手套箱。二甲基甲酰胺和水的混合溶劑(體積比4/1)經(jīng)脫氣處理,在氮?dú)夥諊拢苯愚D(zhuǎn)入微波管中。溶液置于油浴中,在80℃條件下攪拌24小時,再倒入丙酮中產(chǎn)生紅色沉淀。紅色沉淀物經(jīng)過濾、丙酮和甲醇洗滌后,溶于去離子水中,再把水溶液轉(zhuǎn)移到透析袋(MWCO:3500-5000)中透析。透析進(jìn)行1周,每12小時換一次去離子水。然后經(jīng)冷凍干燥處理得深紅色固體 PBTBTz-SO3Na。磺酸側(cè)基使PBTBTz-SO3Na僅溶于水,不溶于其他極性有機(jī)溶劑,因此使用過量的四丁基溴化銨的交換法來處理 PBTBTz-SO3Na以提高其溶解性,得聚合物 PBTBTz-SO3TBA,經(jīng)凝膠滲透色譜(二甲基甲酰胺溶劑)測試(如圖3),得高分子量共軛聚電解質(zhì),其Mn=94 KDa,PDI=2.8。該證明了基于苯并噻二唑和苯并三唑缺電子主鏈結(jié)構(gòu)的共軛聚電解質(zhì)的聚合方法從含離子側(cè)鏈的單體直接聚合成共軛聚電解質(zhì)的方法是可行的。
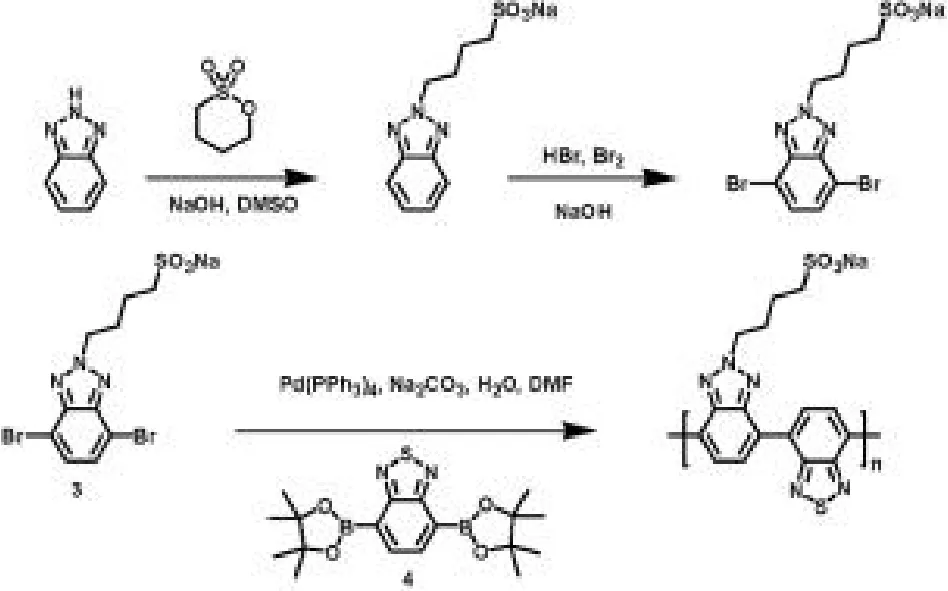
圖 2:PBTBTz-SO3Na的合成
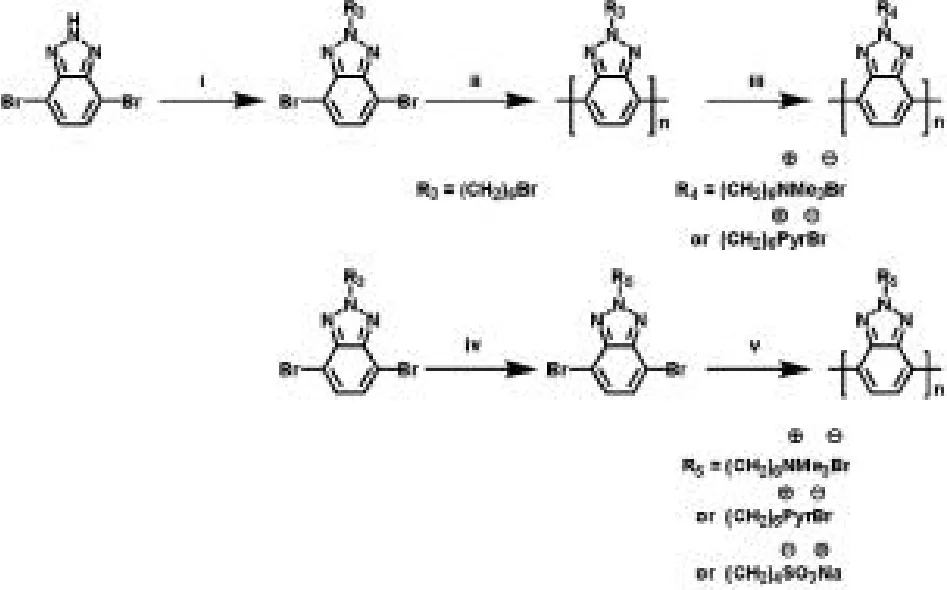
圖3:PBTz-TMABr和PBTz-PyrBr的合成。反應(yīng)條件:(i)4,7-二溴-2H-苯并三唑,1,4-二溴丁烷和氫氧化鉀在60℃條件下回流5 h。(ii)四(三苯基膦)鈀,碳酸鈉,4,7-二溴-2-(6-溴-己基)-2H-苯并三唑在toluene/H2O溶劑中、90℃條件下進(jìn)行24 h。(iii)末端含溴的側(cè)鏈均聚物置于THF/H2O溶劑中,加入三甲胺或者吡啶,在60℃條件下反應(yīng)24小時。(iv)4,7-二溴-2-(6-溴-己基)-2H-苯并三唑4,7-二溴-2-(6-溴-己基)-2H-苯并三唑置于THF/H2O溶劑中,加入三甲胺或者吡啶,在60℃條件下反應(yīng)24小時。(v)將離子末端的單體置于雙(1,5-環(huán)辛二烯)鎳(0),2,2-聯(lián)吡啶和1,5-環(huán)辛二烯中反應(yīng)進(jìn)行48h。
(3)使用 Yamamoto聚合方法合成 PBTz-PyrBr、PBTz-TMABr和 PBTz-SO3Na。
為了進(jìn)一步提高基于苯并三唑和苯并噻二唑主鏈的聚電解質(zhì)的溶解性,含長側(cè)鏈的均聚電解質(zhì)即含離子側(cè)鏈的均聚苯并三唑是最理想的選擇。合成過程如圖 3,PBTz-PyrBr,PBTz-TMABr和PBTz-SO3Na通過從中性聚合物PBTz-Br的官能化反應(yīng)合成時,前體聚合物鏈上的官能團(tuán)的定量轉(zhuǎn)化不完全或者不能反應(yīng),結(jié)果轉(zhuǎn)變?yōu)椴蝗苄缘墓曹椌垭娊赓|(zhì)。Yamamoto聚合法直接將含離子側(cè)鏈官能團(tuán)的單體一步聚合成共軛聚電解質(zhì)PBTz-PyrBr、PBTz-TMABr和PBTz-SO3Na,共軛聚電解質(zhì)的具體合成過程是:在氮?dú)夥諊拢瑢⒌饶柫康亩鍐误w(1.0摩爾比)緩慢滴入雙(1,5-環(huán)辛二烯)鎳(0)(1.0摩爾比)、2,2-聯(lián)吡啶(1.0摩爾比)和1,5-環(huán)辛二烯(1.0摩爾比)的溶液中。溶液在氮?dú)夥諊?5℃條件下攪拌2天,冷卻至室溫后,將混合物倒入氯仿中沉淀,過濾后產(chǎn)物經(jīng)索氏萃取,即用氯仿、丙酮和己烷洗滌,然后溶解在去離子水中,再在透析管(MWCO=3500-5000Da)中進(jìn)行透析反應(yīng),透析進(jìn)行一周,每12小時換一次去離子水。最好經(jīng)冷凍干燥得紅色固體。經(jīng)凝膠滲透色譜(水溶劑,以乙二醇為標(biāo)準(zhǔn))測試,得高分子量的共軛聚電解質(zhì)PBTz-PyrBr、PBTz-TMABr和PBTz-SO3Na,即:PBTz-PyrBr:Mn=112kDa,PDI=3.2,PBTz-TMABr:Mn=157kDa,PDI=1.5,PBTz-SO3Na:Mn=172kDa,PDI=3.7,所得的含吡啶和季胺鹽末端側(cè)鏈的PBTz-PyrBr和PBTz-TMABr可以溶解于甲醇和水中,而含磺酸基末端側(cè)鏈的PBTz-SO3Na只溶于水。說明了通過從中性聚合物PBTz-Br的官能化反應(yīng)合成不能成功的轉(zhuǎn)化為PBTz-PyrBr、PBTz-TMABr和PBTz-SO3Na,只有Yamamoto聚合方法,即基于苯并三唑缺電子主鏈結(jié)構(gòu)的共軛聚電解質(zhì)的聚合方法從帶電荷側(cè)鏈端單元的單體直接聚合到共軛聚電解質(zhì)是可行的。
3 總結(jié)
由于基于苯并噻二唑和苯并三唑缺電子主鏈結(jié)構(gòu)的非離子共軛聚合物的側(cè)鏈官能團(tuán)(即鹵素)不能完全轉(zhuǎn)化為離子官能團(tuán)。因此,我們發(fā)現(xiàn)一種一步合成共軛聚合物的方法,即從含離子側(cè)鏈的單體直接聚合成共軛聚電解質(zhì),本文合成一種基于苯并三唑主鏈的一系列N-型共軛聚電解質(zhì)的合成方法。
