vicK基因ATP結合位點對變異鏈球菌生物學功能的影響
2021-06-03 08:18:52黃珊珊宋秀宇徐巧麗饒慧華張加勤
中國人獸共患病學報 2021年5期
黃珊珊,宋秀宇,徐巧麗,饒慧華,張加勤
變異鏈球菌(Streptococcusmutans,S.mutans)是口腔主要致齲菌之一,至今無特效藥根治齲齒。變異鏈球菌感染口腔后粘附牙齒表面形成生物膜,并在其保護下分解糖類,產生酸代謝產物,引發齲齒[1]。而在致齲的過程中,變異鏈球菌需克服各種生存環境的改變生長,這主要依賴原核生物信號傳導雙組份系統(two-component system,TCS)的調控作用[2]。TCS可參與調節細菌的多種生物學作用,如細胞壁形成、粘附、毒力、氧化應激反應等[3]。S.mutansUA159可編碼14 種TCSs,VicRK雙組分系統為其中之一,與細菌毒力相關,VicK為組氨酸激酶(Histidine Kinase,HK),VicR為可與DNA結合的轉錄因子應答調節蛋白(Response Regulator,RR)[4]。
HATPase_c結構域(CA區)是VicK中具有ATP酶活性的特征性區域,顯示激酶活性,是細菌激酶磷酸化的始動環節。以往人們對VicK的研究大多是敲除整個vicK基因,沒有單獨對HATPase_c結構域的ATP激酶活性進行研究。為研究vicK基因HATPase_c結構域中ATP結合位點對變異鏈球菌的影響,本研究采用同源重組的方法敲除vicK基因ATP結合位點核心區域(命名為ΔvicKATP基因),驗證其蛋白ATP激酶活性,構建補償菌株,觀察vicK基因ATP結合位點對變異鏈球菌生長、產酸耐酸、胞外多糖(exopolysaccharides,EPS)和生物膜形成能力等生物學功能的影響。
1 材料和方法
1.1菌株和質粒 變異鏈球菌UA159由第四軍醫大饋贈;質粒pCrePA和pIB107由堪薩斯大學醫學中心 Biswas Indranil 教授惠贈;pET-His、E.coliDH5、E.coliBL21(DE3)、pET-21(b)、pDLP為前期實驗構建。

1.3實驗步驟
1.3.1vicK基因HATPase_c結構域預測 利用expasy、phyre2和軟件TargetATPsite[5]預測HATPase_c結構域和其中的ATP結合位點,設計引物敲除vicK基因HATPase_c結構域ATP結合位點的核心區域。
1.3.2vicKATP突變同源重組載體構建 以變異鏈球菌UA159基因組DNA為模板,引物見表1,PCR擴增vicK-X1-267片段,將vicK-X1-267片段和pET-His載體分別用BamHI/NheI酶切,T4 DNA連接酶建立連接體系,轉化E.coliDH5感受態細胞,氨芐西林(Ampicillin,Amp)(100 g/mL)篩選陽性菌落,37 ℃過夜培養并抽提質粒獲得重組載體pET-His-vicK-X1-267(pK1)。利用quickchange primer design設計引物,用quick change法構建vicKATP基因突變質粒,同時引入SmalI平末端酶切位點。quick change法建立反應體系:templet(pK1)1 μL(50 ng/μL),5×FastPfu reaction buffer 10 μL,2.5 mmol/L dNTPs 1 μL,FastPfu 0.5 μL,qF1、qR1各2 μL(10 μmol/L),加滅菌水至50 μL。反應條件:95 ℃預變性3 min;95 ℃變性45 s,55~60 ℃退火45 s,68 ℃延伸15 min,95 ℃變性,17 個循環;72 ℃延伸10 min,12 ℃永久。DpnI處理PCR產物,50 μL PCR產物中加入0.5 μLDpnI,37 ℃水浴5 h。由于模板DNA上有甲基化位點,而用PCR新擴增出來的DNA無甲基化修飾,DpnI可識別DNA序列上的甲基化位點,切斷野生型模板DNA,留下突變DNA。測序驗證成功的載體pET-His-vicKATP命名為pK2。SmalI酶切pK2,以質粒pIB107為模板,PCR擴增kan基因盒(lox71-kan-lox66),用PfIu DNA polymeras將lox71-kan-lox66片段補平,連接,轉化。Amp(100 g/mL)和卡那霉素(Kanamycin,Kan)(50 g/mL)篩選陽性菌落,獲得同源重組載體pK2:loxPkan(pK3),經PCR、酶切及測序鑒定成功。

表1 PCR擴增引物
1.3.3UA159 SΔvicKATP突變株構建 參照文獻[6],將pK3經BamHI線性化后,CSP催化轉化UA159,并接種于含Kan(300 g/mL)的Todd-Hewitt Broth plus 0.2%Yeast Extract(THY)固體培養基37 ℃培養24 h,篩選陽性克隆菌落(SΔvicKATP::kan)。同法將熱敏質粒pCrePA轉化SΔvicKATP::kan突變株,pCrePA含emr基因,接種含紅霉素(Erythromycin,Em)(10 g/mL)的THY固體培養基,30 ℃ 48 h篩選陽性菌落,利用Cre酶識別loxP位點剔除kan基因。轉種37 ℃培養基過夜使pCrePA質粒丟失。挑取單個菌落平行接種300 g/mL Kan、10 g/mL Em和無抗性THY固體培養基,37 ℃培養24 h。篩選Kanr和Emr平板不生長,無抗性平板上生長的菌落,即無標記SΔvicKATP突變株。測序鑒定。
1.3.4UA159vicKATP補償株構建 以UA159基因組DNA為模版,PCR擴增vicK基因開放閱讀框。vicK片段和穿梭表達質粒pDLP分別用NdeI/KpnI酶切,連接,轉化,Kan(50 g/mL)LB平板篩選陽性菌落。所獲質粒經PCR和酶切鑒定成功即為補償質粒pDLP-vicK(pK4)。補償質粒轉化SΔvicKATP突變株,方法和步驟同上所述,含300 μg/mL KanrTHY平板篩選并獲得vicKATP補償株為SvicK。
1.3.5RT-PCR驗證vicK基因轉錄 提取UA159、SΔvicKATP突變株和SvicK補償株RNA,合成第一鏈cDNA,PCR擴增cDNA的vicK產物。驗證SΔvicKATP突變株和SvicK補償株的vicK基因轉錄活性。
1.3.6vicK基因HATPase_c結構域缺失蛋白表達純化和ATP酶活性檢測 分別以UA159和SΔvicKATP突變株基因組DNA為模版,PCR擴增vicK基因開放閱讀框。BamHI和HindIII雙酶切表達載體pET-21(b)、vicK基因和vicKATP基因片段,連接,轉化,Ampr LB平板篩選陽性菌落。所獲質粒經PCR和酶切鑒定成功即為表達載體pET-21(b)-vicK和pET-21(b)-ΔvicKATP,轉化感受態細胞E.coliBL21(DE3)獲得pET-21(b)-vicK-BL21和pET-21(b)-ΔvicKATP-BL21,IPTG誘導蛋白表達,Ni-NTA SefinoseTM純化蛋白。目的蛋白經SDS-PAGE電泳分析初步鑒定,-80 ℃保存備用。
1.3.7細菌培養 生物學功能實驗均將UA159、SΔvicKATP突變株和SvicK補償株在37 ℃、5% CO2條件下靜置過夜培養。
1.3.8生長速率 將菌株分別以1∶100比例接種50 mL THY液體培養基,37 ℃靜置培養。每隔1 h讀取OD600值,對數生長期(OD600≈0.5)每隔30 min讀取1次數值。重復3次實驗取平均值,記錄數值并作曲線圖。
1.3.9產酸實驗 將菌株分別以1∶20比例接種THY培養基,37 ℃靜置培養。每隔1 h測量pH值,分別為0、1、2、3、4、5、6 h。重復3次實驗取平均值,記錄數值并繪制曲線圖。
1.3.10酸耐受實驗 分別取菌株1 mL,生理鹽水洗滌。加200 μL 0.05 mol/L甘氨酸-鹽酸緩沖液(pH2.8),分別反應60 min、90 min,各取100 μL 菌液梯度稀釋鋪板。置37 ℃培養48 h。菌落計數,重復3 次實驗取平均值,并繪圖。
1.3.11生物膜培養 將菌株轉種到THY培養基中培養至對數生長期(OD600≈0.5),以1∶100比例接種于50 mL含0.5%蔗糖的THY培養基,取1 mL 稀釋物注入無菌的十二孔平底培養板中,并分別放入一張無菌蓋玻片,37 ℃ CO2培養箱靜置培養48 h,洗滌培養板去除未粘附細菌,風干,每孔加入250 μL 0.1%結晶紫溶液覆蓋孔底,室溫染色5 min,水洗多余染液并風干,鏡下觀察生物膜。1 mL(乙醇∶丙酮=8∶2)的萃取液萃取生物膜,酶標儀檢測OD570吸光度值。
1.3.12掃描電鏡(SEM)觀察生物膜及EPS形成情況 生物膜培養后,取出硅片,PBS吹打洗滌細菌,除去表面生物膜的殘留雜質,2.5%戊二醛固定10~20 min,30%、50%、70%、90%、100%乙醇各10~20 min干燥杯中脫水,再從乙醇逐步過渡到叔丁醇,干燥杯4 ℃貯存,電鏡室冷凍干燥,噴金后電鏡觀察。
1.4統計學分析 數據采用Graphpad Prism 8.0軟件繪圖分析,應用雙因素方差分析(two-way ANOVA)比較實驗組和對照組ATP激酶活性、產酸、酸耐受差異,應用單因素方差分析非參數檢驗(Kruskal-Wallis test)比較實驗組和對照組生物膜形成差異。檢驗水準α=0.05。
2 結 果
2.1vicK基因HATPase_c結構域預測vicK基因HATPase_c結構域和其ATP結合位點如圖1所示。設計引物對ATP結合位點核心區域396-402 aa進行缺失突變,vicK基因HATPase_c結構域和其ATP結合位點位置推斷突變位置不影響vicK基因其他功能結構域的表達。
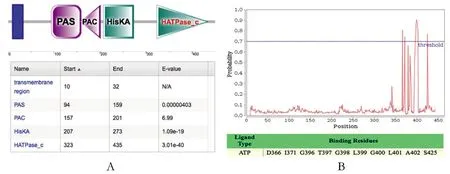
A:HATPase_c結構域預測;B:ATP結合位點預測
2.2UA159 SΔvicKATP突變株構建與鑒定 構建成功的同源重組載體pK3經BamHI線性化后轉化UA159,在Kanr平板上生長說明lox71-kan-lox66已經成功整合到UA159基因組。熱敏質粒pCrePA轉化ΔvicKATP::kan突變株,利用Cre酶識別loxP位點可剔除kan基因。37 ℃使pCrePA質粒丟失。突變株在Kanr和EmrTHY平板上不生長,只在無抗性THY平板上生長,驗證kan基因和pCrePA質粒均成功去除,成功構建無標記SΔvicKATP突變株。SΔvicKATP突變株測序結果顯示vicK基因的ATP結合位點序列已缺失,只存留38 bp的loxP位點(圖2)。
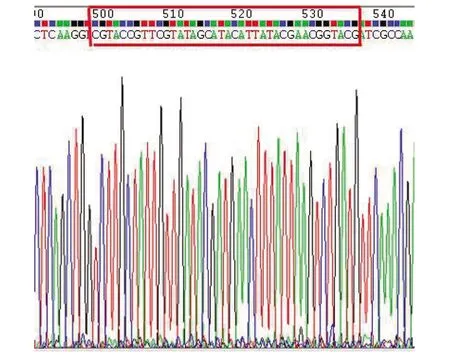
紅色方框內為刪除kan基因后殘留38 bp的loxP位點
2.3UA159vicK補償株構建與鑒定 將構建成功的補償質粒pDLP-vicK(pK4)轉化vicKATP突變株,Kanr的THY平板篩選獲得vicK補償株。由于SΔvicKATP突變株刪除了ATP結合位點核心區域21 bp,引入lox71-kan-lox66,而Cre重組酶識別loxP位點刪除kan基因后殘留兩端38 bp的loxP位點,用PCR擴增vicK基因和突變基因產物大小區別不大,無法區分驗證突變株和補償株,且無法確定補償vicK基因是否有轉錄活性。因此,對陽性轉化SvicK補償株和SΔvicKATP突變株vicK基因轉錄情況用RT-PCR驗證。結果如下圖所示,SΔvicKATP突變株RT-PCR產物電泳結果無相關條帶出現,進一步說明SΔvicKATP突變株構建成功,SvicK補償株vicK基因有轉錄活性(圖3)。
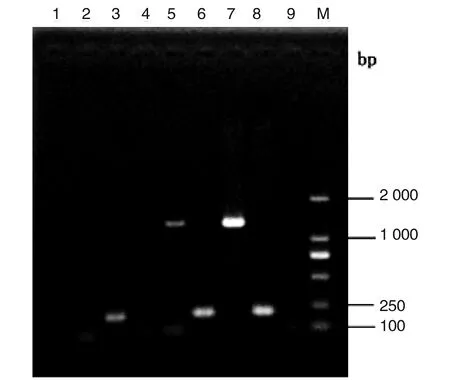
1:突變株SΔvicKATP RNA的RT-PCR擴增vicK產物;2:突變株SΔvicKATP cDNA 的RT-PCR擴增vicK產物;3:突變株SΔvicKATP cDNA 的RT-PCR擴增gyrA內參產物;4:補償株SvicK RNA的RT-PCR擴增vicK產物;5:補償株SvicK cDNA的RT-PCR擴增vicK產物;6:補償株SvicK cDNA的RT-PCR擴增gyrA內參產物;7:UA159野生株基因組的PCR擴增vicK產物;8:UA159野生株基因組的PCR擴增gyrA內參產物;9:陰性對照;M:DNA Marker DL2000
2.4vicK基因HATPase_c結構域缺失蛋白表達純化和ATP酶活性檢測 經IPTG誘導表達目的蛋白,ΔVicKATP蛋白為45.7 kDa,VicK蛋白為50 kDa。Ni-NTA SefinoseTM純化蛋白,imidazole濃度400 mmol/L洗脫目的蛋白(圖4)。檢測ΔVicKATP蛋白和VicK蛋白的ATP激酶活性,繪制標準曲線(圖5A)。結果提示隨ATP濃度增加,發光強度增加。蛋白ATP酶活性越高,結合ATP能力越強,導致游離ATP減少,發光強度降低,即蛋白ATP酶活性與發光強度成反比。隨著兩種蛋白摩爾濃度增加,VicK蛋白發光強度呈明顯降低趨勢,而ΔVicKATP蛋白發光強度無明顯變化,差異有統計學意義(FVicK-ΔVicKATP=22.87,P<0.05)(圖5B)。結果顯示,VicK蛋白的ATP激酶活性明顯高于ΔVicKATP蛋白,ΔVicKATP蛋白幾乎失去ATP激酶活性。驗證了SΔvicKATP突變株構建成功,缺失了ATP的結合能力。
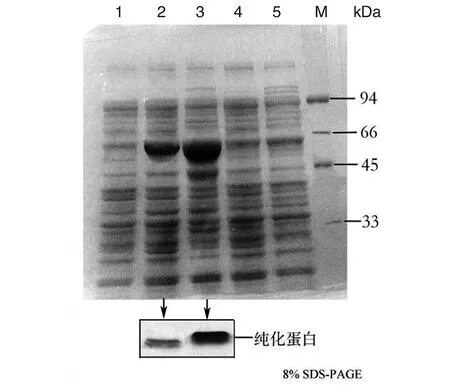
1:對照株E.coli BL21(DE3)裂解上清液;2: pET-21(b)-ΔvicKATP-BL21裂解上清液及純化蛋白;3: pET-21(b)-vicK-BL21裂解上清液及純化蛋白;4:pET-21(b)-ΔvicKATP-BL21裂解沉淀物;5:pET-21(b)-vicK-BL21裂解沉淀物;M:蛋白質marker
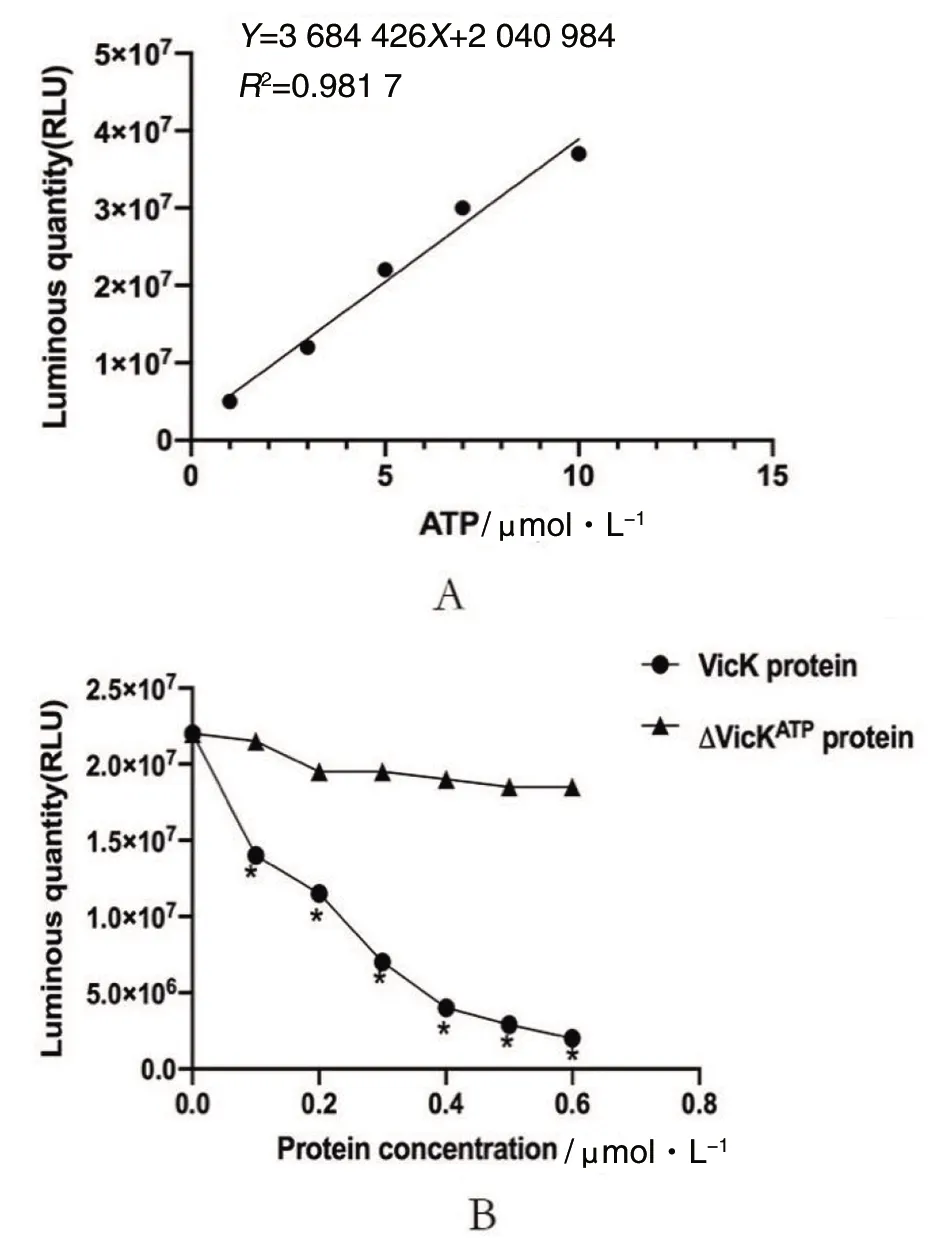
A:標準曲線;B:不同摩爾濃度ΔVicKATP蛋白和VicK蛋白ATP激酶活性;*P<0.05,compared with ΔVicKATP protein
2.5生長曲線 每隔60 min測量OD600。與野生株UA159比較,SΔvicKATP突變株生長較為緩慢,但穩定期基本可達到同一水平,而SvicK補償株基本恢復至野生株的生長水平,見圖6。
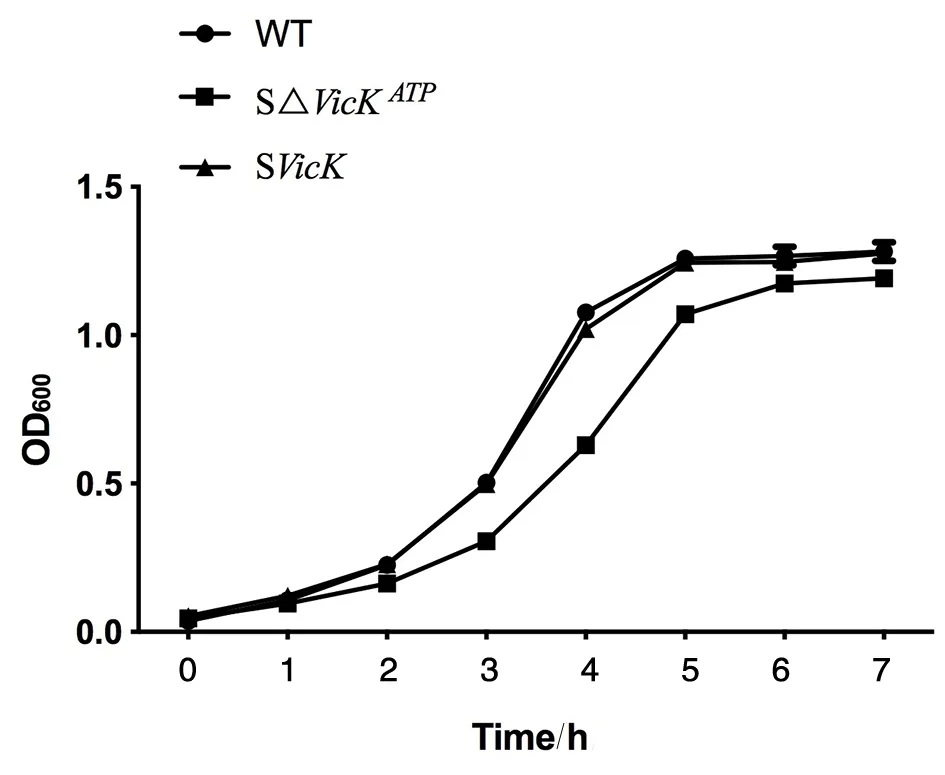
圖6 UA159、SΔvicKATP突變株和SvicK補償株的生長曲線
2.6產酸實驗 相比野生株,隨時間增加SΔvicKATP突變株PH值下降更緩慢,即突變株產酸量減少,差異有統計學意義(q1、2、3、4、5、6:WT-SΔvicKATP=7、8.875、18.35、40.83、99.03、10,P<0.05)。SvicK補償株pH值下降也較野生株緩慢,產酸量稍減少,部分差異有統計學意義(q2、3、4、5:WT-SvicK=13、61、116、164,P<0.05),但沒有突變株明顯,說明ATP結合位點突變使菌株產酸量減少,補償株不能完全恢復到野生株水平(圖7)。
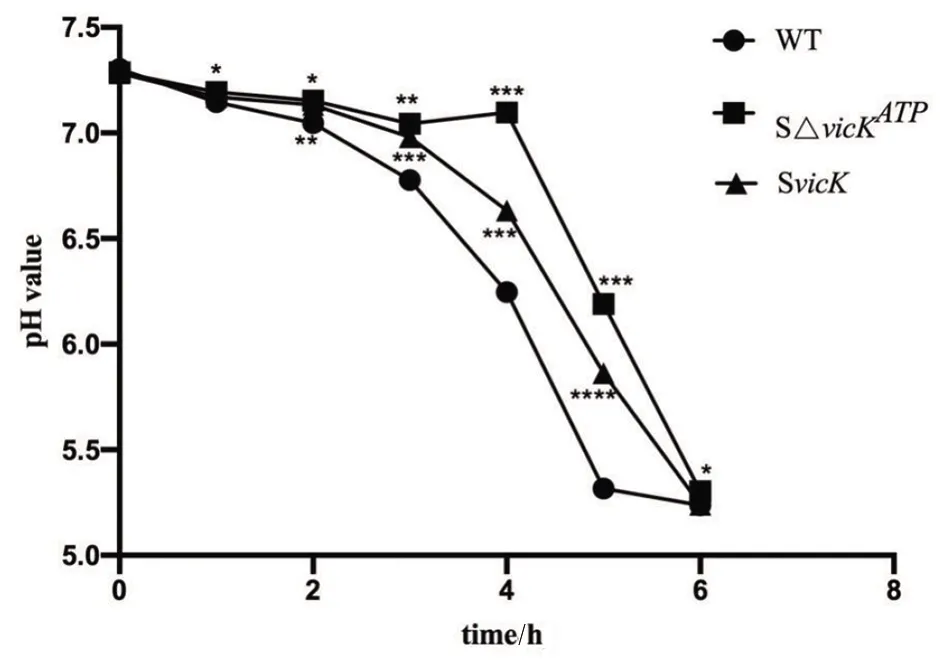
*P<0.05,**P<0.01,***P<0.005,****P<0.001,compared with WT
2.7酸耐受實驗 在致死性酸性環境中(pH 2.8),隨著酸處理時間延長,UA159、SΔvicKATP突變株、SvicK補償株生存率呈下降趨勢,但與野生株相比,SΔvicKATP突變株反而下降的更少,差異有統計學意義(q60:WT-90:WT=45.20,q60:WT-90:SΔvicKATP=14.14,q60:WT-90: SvicK=34.96,P<0.05),提示突變株耐酸性增高。90 min時,補償株和野生株差異有統計學意義(q90:WT-SvicK=23.03,P<0.05),提示隨酸處理時間增加,ATP結合位點突變對酸耐受性增加(圖8)。
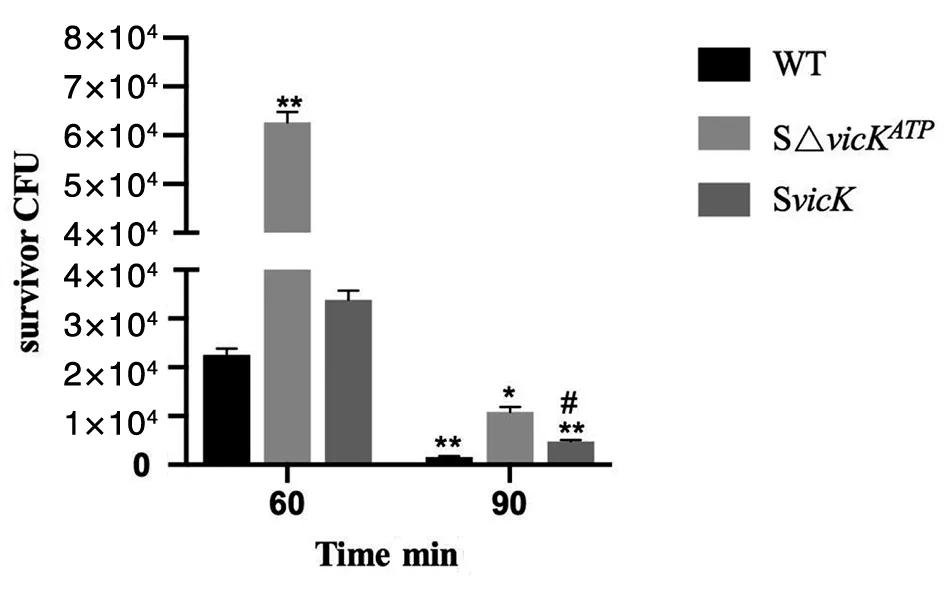
*P<0.05,**P<0.01,compared with WT60;#P<0.05, compared with WT90
2.8vicK基因ATP結合位點突變對變異鏈球菌生物膜及EPS形成的影響 與野生株相比,SΔvicKATP突變株生物膜形成明顯減少,粘附力降低,且致密性降低,補償株SvicK生物膜形成能力基本與野生株相當(圖9)。用配制的溶液萃取生物膜,將萃取液于OD570檢測,結果與結晶紫染色一致:突變株與野生株比較,差異有統計學意義(Z=2.695,P<0.05),說明SΔvicKATP突變株使變異鏈球菌生物膜形成能力降低。補償株與野生株比較差異無統計學意義,從圖9中看出補償株起到了部分補償作用,但還是有一定差異。電鏡SEM結果觀察到:SΔvicKATP突變株玻片上的生物膜比野生株薄,與生物膜培養結晶紫染色結果一致,細菌量比野生株少,EPS生成量相比野生株明顯減少,補償株幾乎恢復到野生株水平(圖10)。
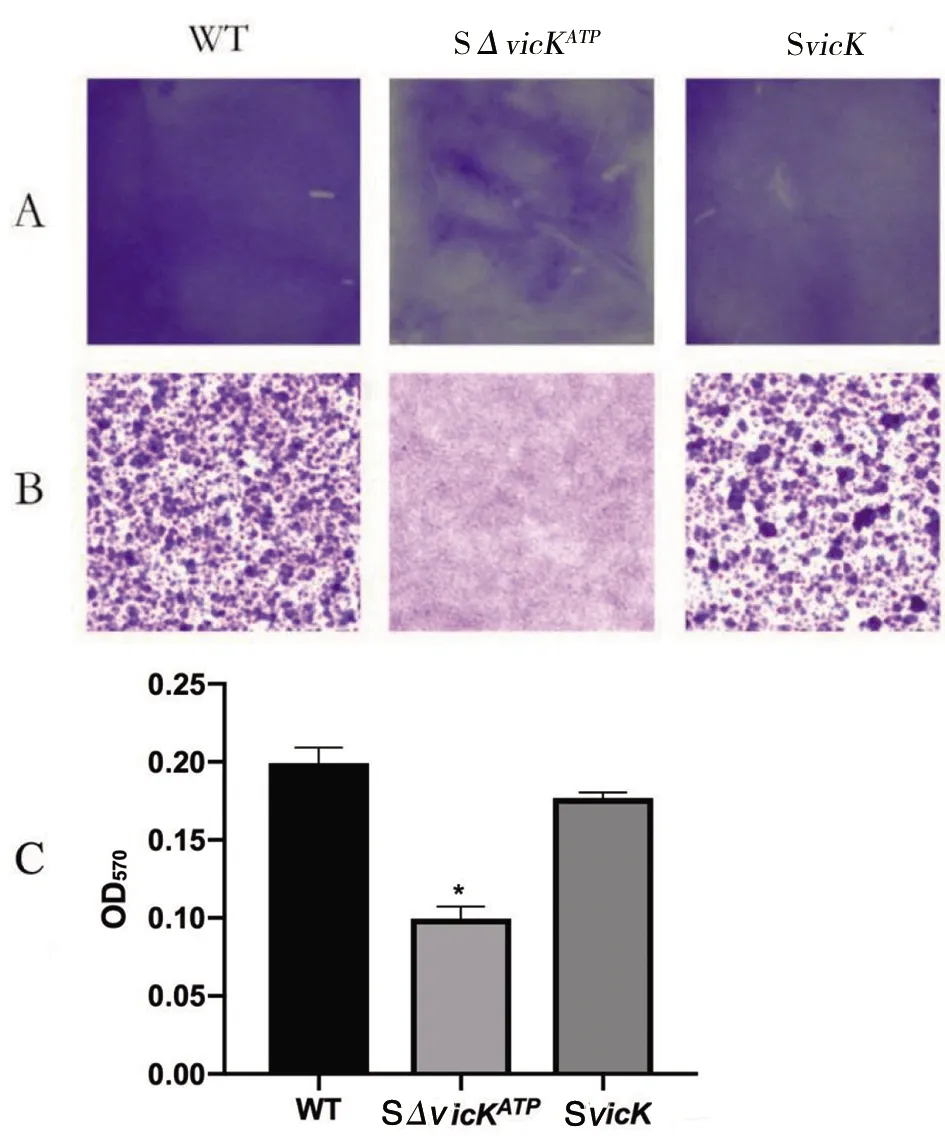
A:生物膜形成;B:光學顯微鏡下生物膜形態(100倍);C:生物膜萃取液OD570值;*P<0.05,compared with WT

A:50 000倍;B:5 000倍
3 討 論
TCS由位于膜上的組氨酸激酶受體HK和位于胞內的調節子RR組成[7]。HK感受外界信號并發生自磷酸化,將磷酸基團轉移到RR上將其磷酸化,調控下游目的基因表達[3,8]。HK由N端傳感器復合體和C端催化中心組成。N端包括胞外感受區、跨膜區(TM)和胞內感受區(HAMP、PAS)[9]。C端包括DHp和HATPase_c結構域(CA區)。HATPase_c結構域具有催化活性并含有 ATP 結合位點,識別結合ATP,并呈遞磷酸基團給DHp區的His217,使其發生自身磷酸化,是細菌激酶磷酸化的始動環節[10]。細菌HATPase_c結構域在人體中沒有發現,近年來已成為抗菌藥物研究的重要靶點[11]。研究表明雙組分系統主要存在于細菌、一些低等真核生物和一些原生動物中,在人體真核細胞中卻不存在[12],因此可通過研究雙組分系統蛋白毒力調節機制,尋找變異鏈球菌雙組分系統中的藥物新靶點,特異高效地抑制病原菌而對宿主不造成影響。
變異鏈球菌中的雙組分VicRK系統的vicK基因,其編碼的組氨酸激酶VicK感受外界刺激信號并傳遞給應答調節蛋白VicR,開始信號通路轉導,從而調節相關蛋白的轉錄翻譯[13]。因此,vicK基因在變異鏈球菌調節毒力機制作用的研究至關重要。研究表明,vicK基因對變異鏈球菌的生長、產酸、耐酸、粘附力、氧化應激、生物膜和糖酵解均有調控作用,對產生EPS研究較少,EPS對細菌生物膜形成和毒力均有重要作用[2,4,14-16]。這些大多是敲除整個vicK基因的研究結果。但VicRK是復雜的雙組分調控系統,VicK不僅有組氨酸激酶活性,還有磷酸酶和磷酸轉移酶活性。為研究vicK基因HATPase_c結構域中ATP結合位點激酶磷酸化功能對變異鏈球菌的影響,本研究通過敲除vicK基因ATP結合位點,觀察vicK基因ATP結合位點對變異鏈球菌生物學功能的影響。研究表明,VicK蛋白的保守氨基酸殘基P222、T221和A439有磷酸酶活性,其中P222、T221點突變,VicK蛋白失去磷酸酶活性,A439位點缺失突變,VicK蛋白磷酸酶活性顯著降低;A439和W443有磷酸轉移酶活性,突變后VicK蛋白磷酸轉移酶活性顯著降低10。本研究中對HATPase_c結構域中ATP結合位點核心區域396-402 aa進行缺失突變,突變位點不涉及已知磷酸酶和磷酸轉移酶活性位點,保留了磷酸酶和磷酸轉移酶活性。蛋白結構預測分析發現,VicK蛋白氨基酸全長450 aa,HATPase_c結構域位于C端323-435 aa,突變位置不影響vicK基因其他功能結構域的表達。
為研究vicK基因ATP結合位點對變異鏈球菌生物學功能的影響,本研究用基因克隆和quick change法成功構建了vicKATP基因突變同源重組表達載體。利用Cre-loxP系統和同源重組法將ATP結合位點突變的目的基因整合到變異鏈球菌的基因組中,穩定表達突變目的基因,成功構建SΔvicKATP突變株。利用Cre酶識別并刪除引入的lox71-kan-lox66抗性基因盒,構建無標記的SΔvicKATP突變株。同時,克隆vicK片段,采用穿梭表達質粒pDLP轉化SΔvicKATP突變株,成功構建具有vicK基因轉錄活性的補償株SvicK。成功構建VicKATP蛋白表達株,表達純化蛋白,并檢測其幾乎失去ATP酶活性,驗證了SΔvicKATP突變株構建成功,缺失了ATP的結合能力。通過描繪生長曲線、產酸、酸耐受、EPS和生物膜等生物學功能實驗,研究vicK基因ATP結合位點對變異鏈球菌的影響。研究結果顯示:與野生株UA159比較,SΔvicKATP突變株生長和產酸都受到抑制,酸耐受性增強,SvicK補償株不能完全恢復到野生株水平;生物膜通過結晶紫染色、電鏡SEM觀察,SΔvicKATP突變株生物膜形成明顯減少,粘附力降低,且致密性降低,生物膜形成能力降低,補償株SvicK生物膜形成能力基本與野生株相當,其中SEM還觀察到SΔvicKATP突變株EPS生成量相比野生株明顯減少。實驗結果與S.mutansUA159的vicK基因全敲除結果大致相同,揭示vicK基因HATPase_c結構域中ATP結合位點對變異鏈球菌生物學功能和毒力機制有重要影響,具體機制待進一步研究。
變異鏈球菌vicKATP基因的成功敲除及生物學功能研究,為進一步研究變異鏈球菌vicK基因ATP結合位點表達調控毒力機制提供了無標記基因缺陷株模型和實驗依據,為研發抗菌藥物奠定基礎。
利益沖突:無
