基于UPLC的疏風解毒膠囊指紋圖譜及多成分含量測定
2021-06-11 06:51:06金蕾張貴萍王溫才
中國現代中藥 2021年4期
金蕾,張貴萍,王溫才
1.青島市市立醫院 藥劑科中心藥房,山東 青島 266000;2.青島市市立醫院 藥物臨床試驗研究科,山東 青島 266000
疏風解毒膠囊由虎杖、連翹、板藍根、柴胡、敗醬草、馬鞭草、蘆根、甘草8味中藥制成,具有疏風清熱、解毒利咽的功效,用于急性上呼吸道感染屬風熱證,癥見發熱、惡風、咽痛、頭痛、鼻塞、流濁涕、咳嗽等[1]1784。現代藥理學研究表明,疏風解毒膠囊對細菌、病毒、真菌等具有廣泛的抑制作用[2],臨床廣泛用于治療社區獲得性肺炎、手足口病、帶狀皰疹、慢性支氣管炎、上呼吸道感染、流感等各類疾病,且治療效果較為明顯[3]。方中虎杖利濕退黃、清熱解毒、散瘀止痛、止咳化痰,《中華人民共和國藥典》(以下簡稱《中國藥典》)2020年版含量測定指標成分為大黃素和虎杖苷[1]217;連翹清熱解毒、消腫散結、疏散風熱[4],《中國藥典》2020年版含量測定指標成分為連翹苷和連翹酯苷A[1]177;板藍根性寒,清熱解毒、涼血利咽[5],《中國藥典》2020年版含量測定指標成分為(R,S)-告依春[1]214;柴胡疏散退熱、疏肝解郁、升舉陽氣,《中國藥典》2020年版含量測定指標成分為柴胡皂苷a、柴胡皂苷d[1]293;馬鞭草活血散瘀、解毒、利水、退黃、截瘧,《中國藥典》2020年版含量測定指標成分為熊果酸、齊墩果酸[1]53。疏風解毒膠囊現收載于《中國藥典》2020年版,標準以連翹中連翹苷和虎杖中虎杖苷為質量控制指標成分[1]1784,目前,僅有對其單個或多成分含量測定的文獻報道[6-8],未見其指紋圖譜的文獻報道。為了能全面反映疏風解毒膠囊的質量特性,本研究建立疏風解毒膠囊超高效液相色譜法(UPLC)指紋圖譜,并同時測定 9個指標成分的含量。
1 材料
1290型超高效液相色譜儀、二極管陣列檢測器(安捷倫科技有限公司);DHG-9246A型電熱恒溫鼓風干燥箱(上海精宏實驗設備有限公司);XS205DU型電子天平(d=0.01 mg,瑞士梅特勒公司);FRQ-1006HTD型超聲波清洗機(杭州法蘭特超聲波科技有限公司);Option-Q型純水機(ELGA公司)。
疏風解毒膠囊均為同一生產廠家(市售),12批樣品批號分別為1852144、1912411、1906521、1903624、1812142、2002141、2003512、1906321、0911225、2001423、2003214、2004711,編號分別為S1~S12,規格為0.52 g/粒。對照品(R,S)-告依春(批號:111753-201706,純度:100.0%)、連翹苷(批號:110821-201816,純度:95.1%)、連翹酯苷A(批號:11810-201707,純度:97.2%)、柴胡皂苷a(批號:110777-201711,純度:91.1%)、柴胡皂苷d(批號:110778-201711,純度:93.2%)、虎杖苷(批號:111575-201603,純度:87.3%)、大黃素(批號:110756-201913,純度:96.0%)、熊果酸(批號:110742-201823,純度:99.9%)、齊墩果酸(批號:110709-201808,純度:91.1%)均購自中國食品藥品檢定研究院;甲醇、乙腈(德國Merck公司,色譜純);水為娃哈哈純凈水。
2 方法與結果
2.1 色譜條件
色譜柱:Agilent XDB-C18(100 mm×2.1 mm,1.8 μm);流動相:乙腈-0.05%磷酸溶液(B),梯度洗脫(0~5 min,10%A;5~15 min,10%~25%A;15~22 min,25%~30%A;22~33 min,30%~50%A;33~35 min,50%~10%A);流速:0.3 mL·min-1;檢測波長:0~8 min,220 nm[(R,S)-告依春]、8~13 min,310 nm(虎杖苷、連翹酯苷A)、13~35 min,220 nm(柴胡皂苷a、柴胡皂苷d、連翹苷、大黃素、齊墩果酸、熊果酸);柱溫:30.0 ℃;進樣量:1.00 μL。
2.2 溶液的制備
2.2.1混合對照品溶液 分別取(R,S)-告依春、虎杖苷、連翹酯苷A、柴胡皂苷a、柴胡皂苷d、齊墩果酸、熊果酸、連翹苷、大黃素對照品各適量,精密稱定后加甲醇適量,超聲使溶解(功率:300 W,頻率:40 kHz),配制成含(R,S)-告依春146.9 μg·mL-1、虎杖苷2 285.0 μg·mL-1、連翹酯苷A 684.0 μg·mL-1、柴胡皂苷a 667.0 μg·mL-1、柴胡皂苷d 660.3 μg·mL-1、連翹苷429.7 μg·mL-1、大黃素1 066.0 μg·mL-1、齊墩果酸298.1 μg·mL-1、熊果酸415.0 μg·mL-1的混合對照品儲備液,精密量取對照品儲備液適量,置25 mL棕色量瓶中,用甲醇稀釋至刻度,搖勻,即為混合對照品溶液。
2.2.2供試品溶液 取本品10粒內容物混勻,研細,精密稱定2.0 g,置于具塞燒瓶中,加入75%甲醇50 mL,精密稱定后加熱回流 1 h,取出,放冷,用75%甲醇補足減失質量,搖勻,濾過,即得。
2.3 高效液相色譜法(HPLC)指紋圖譜的建立及方法學驗證
2.3.1精密度試驗 取樣品(S1)的供試品溶液,按2.1項下色譜條件,連續進樣5次,記錄色譜峰,10號峰為參照峰(柴胡皂苷a),以各共有峰的相對峰面積和相對保留時間的RSD為考察指標,結果均小于2.0%。
2.3.2穩定性試驗 取供試品溶液(S1),按2.1項下色譜條件,分別在 0、4、8、12、24、36 h進樣,記錄色譜圖,以10號峰為參照峰(柴胡皂苷a),以各共有峰的相對峰面積和相對保留時間的RSD為考察指標,結果均小于2.0%。
2.3.3重復性試驗 取樣品(S1)6份,按2.2.2項下方法制備供試品溶液,按2.1項下色譜條件進樣測定,記錄色譜圖中各待測組分的峰面積和保留時間,以10號峰為參照峰(柴胡皂苷a),以各共有峰的相對峰面積和相對保留時間的RSD為考察指標,結果均小于2.0%。
2.3.4指紋圖譜的建立及相似度分析 取樣品(S1~S10),依照2.2.2項下方法分別制備12批供試品溶液,按2.1項下色譜條件分別進樣,記錄色譜數據,將12批樣品的色譜數據的DCF文件導入“中藥色譜指紋圖譜相似度評價系統”(2012年版),對12批樣品的 UPLC 圖譜進行分析,生成疏風解毒膠囊指紋圖譜,見圖1(A、B)。12批樣品與對照圖譜的相似度分別為0.987、0.979、0.965、0.959、0.976、0.981、0.990、0.953、0.978、0.989、0.964、0.971,其相似度均大于0.95,疏風解毒膠囊同一廠家各批次間相似度良好,質量相對一致。

注:A.指紋圖譜;B.對照指紋圖譜;C.混合對照品色譜圖;D.樣品色譜圖;4.(R,S)-告依春;5.虎杖苷;7.連翹酯苷A;10.柴胡皂苷a;13.柴胡皂苷d;17.連翹苷;19.大黃素;21.齊墩果酸;22.熊果酸。圖1 12批疏風解毒膠囊UPLC圖
2.3.5共有峰指認 采用“中藥色譜指紋圖譜相似度評價系統(2012年版)”對12批樣品的UPLC圖譜進行分析,12批樣品共有22個共有峰,在22個共有峰中,通過與各對照品峰的紫外吸收光譜圖和保留時間比對,指認出4號峰為(R,S)-告依春、5號峰為虎杖苷、7號峰為連翹酯苷A、10號峰為柴胡皂苷a、13號峰為柴胡皂苷d、17號峰為連翹苷、19號峰為大黃素、21號峰為齊墩果酸、22號峰為熊果酸。
2.4 多指標成分的含量測定
2.4.1系統適用性試驗 按2.1項下色譜條件,分別精密吸取2.2項下溶液各1.00 μL進樣,結果各待測成分色譜峰與其相鄰色譜峰分離度均符合要求(>1.5),對稱因子為0.97~1.32,理論塔板數以(R,S)-告依春、虎杖苷、連翹酯苷A、柴胡皂苷a、柴胡皂苷d、連翹苷、大黃素、齊墩果酸、熊果酸峰計均在8900以上。色譜圖見圖1(C、D)。
2.4.2線性關系考察 分別精密吸取2.2.1項下混合對照品儲備液適量,制備系列質量濃度的混合對照品溶液,按2.2項下色譜條件,精密吸取上述系列混合對照品溶液各1.00 μL進樣,記錄色譜圖。以質量濃度為橫坐標,峰面積為縱坐標,繪制標準曲線。結果見表1。

表1 疏風解毒膠囊各成分線性關系考察結果
2.4.3精密度試驗 精密吸取2.2.1項下溶液1.00 μL,按2.1色譜條件重復進樣6次,記錄峰面積。結果表明,(R,S)-告依春、虎杖苷、連翹酯苷A、柴胡皂苷a、柴胡皂苷d、連翹苷、大黃素、齊墩果酸、熊果酸峰面積的RSD分別0.68%、0.93%、0.58%、1.0%、0.69%、0.32%、0.61%、0.78%、0.93%(n=6),表明儀器精密度良好。
2.4.4穩定性試驗 取樣品(S1),按2.2.2項下方法制備供試品溶液,分別于制備后0、4、8、12、24、36 h按2.1項下色譜條件進樣測定,記錄峰面積。(R,S)-告依春、虎杖苷、連翹酯苷A、柴胡皂苷a、柴胡皂苷d、連翹苷、大黃素、齊墩果酸、熊果酸峰面積的RSD分別為1.2%、0.96%、1.0%、0.86%、1.1%、1.3%、0.92%、0.87%、0.95%(n=6),表明在36 h內供試品溶液穩定性較好。
2.4.5重復性試驗 取樣品(S1)6份,按2.2.2項下方法制備供試品溶液,再按2.1項下色譜條件進樣,記錄色譜圖中各待測組分的峰面積,計算得(R,S)-告依春、虎杖苷、連翹酯苷A、柴胡皂苷a、柴胡皂苷d、連翹苷、大黃素、齊墩果酸、熊果酸的平均質量分數分別為0.442、6.891、2.023、2.114、1.970、1.310、3.258、1.604、1.987 mg·g-1,RSD分別0.95%、0.49%、0.87%、1.00%、1.10%、0.69%、0.93%、1.30%、0.97%(n=6),表明方法重復性良好。
2.4.6加樣回收率試驗 精密稱定已知含量的疏風解毒膠囊(S1)細粉6份,每份約1.0 g,分別精密加入混合對照品儲備液3.0 mL,按2.2.2項下方法制備,按2.1項下色譜條件分析,記錄色譜圖中各待測組分的峰面積,計算各成分平均回收率及其RSD。結果見表2。

表2 疏風解毒膠囊9個成分的加樣回收率試驗結果
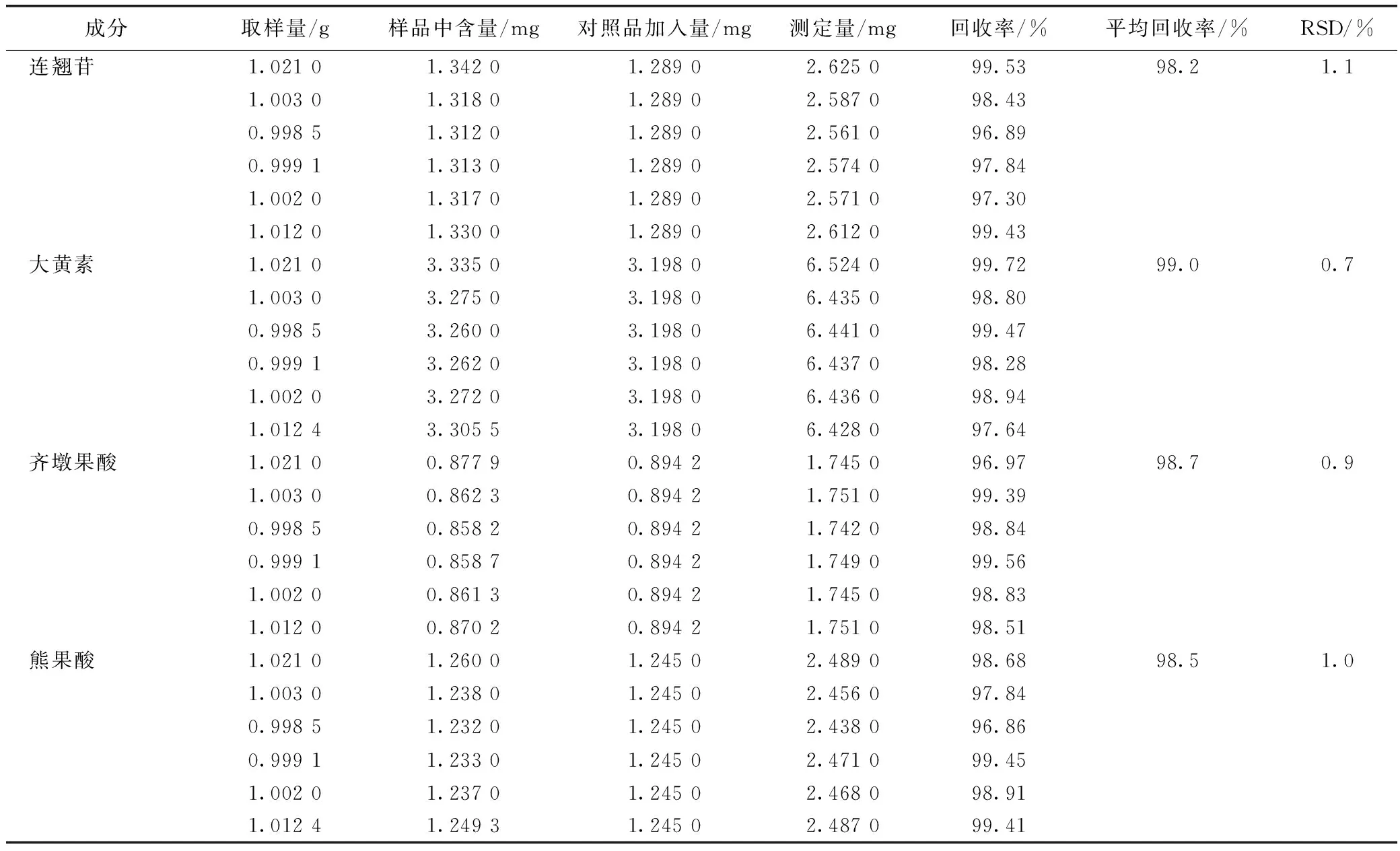
續表2
2.4.7樣品測定 取疏風解毒膠囊12批,按照2.2.2項下方法,平行制備3份,按2.1項下色譜條件測定,記錄(R,S)-告依春、虎杖苷、連翹酯苷A、柴胡皂苷a、柴胡皂苷d、連翹苷、大黃素、齊墩果酸、熊果酸的峰面積,計算上述9個成分含量。結果見表3。

表3 疏風解毒膠囊中9個成分含量測定結果(n=3) mg·g-1
3 討論
3.1 色譜條件的優化
本試驗對不同廠家色譜柱、流動相體系、檢測波長進行考察,選擇Agilent XDB-C18色譜柱(100 mm×2.1 mm,1.8 μm)、AQUITY UPLC BEH C18色譜柱(100 mm×2.1 mm,1.8 μm)、LunaOmega C18色譜柱(50 mm×2.1mm,1.6 μm)對樣品進行色譜分析,發現使用Agilent XDB-C18色譜柱(100 mm×2.1 mm,1.8 μm)分離色譜圖較好;考察甲醇-水、乙腈-水、乙腈-0.05%磷酸溶液等流動相的色譜分離效果,結果以乙腈-0.05%為流動相,色譜峰峰形尖銳,分離效果最佳,基線平穩,保留時間適中;將(R,S)-告依春、虎杖苷、連翹酯苷A、柴胡皂苷a、柴胡皂苷d、連翹苷、大黃素、齊墩果酸、熊果酸的對照品溶液分別注入液相色譜儀,在200~400 nm進行全波長掃描,結果最大吸收波長分別為 245 nm[(R,S)-告依春]、306 nm(虎杖苷)、330 nm(連翹酯苷A)、210 nm(柴胡皂苷a、柴胡皂苷d)、277 nm(連翹苷)、254 nm(大黃素)、210 nm(齊墩果酸、熊果酸)。根據各檢測成分紫外吸收的吸收峰特征,選擇不同時段220、310 nm 2波長切換法進行測定,峰形相對較好,包含色譜信息較多,因此,確定檢測波長為220、310 nm。
3.2 提取方式的優化
預實驗比較了不同提取溶劑的提取效果,以不同體積分數乙醇(100%、75%、30%)和水為提取溶劑時,色譜峰基線出現漂移現象,基線不平穩;以甲醇(100%、75%、30%)為提取溶劑時,75%甲醇對各組分的提取效率較高。提取方法的考察,預實驗比較了超聲提取法和加熱回流法對各組分提取效果的差異,結果加熱回流提取法對各組分的提取較充分,特別是虎杖苷、大黃素、齊墩果酸、熊果酸4個成分含量差別較明顯,為了使提取完全,故最終采用加熱回流作為提取法。
3.3 樣品測定結果分析
中成藥化學成分較復雜,其療效不是多個化學成分藥用作用的簡單相加,指紋圖譜技術可對中藥制劑進行整體性評價,對制劑質量控制有針對性和合理性[9-12]。本研究采用“中藥色譜指紋圖譜相似度評價系統”(2012年版)建立了該制劑指紋圖譜,12批樣品相似度均大于0.95,表明疏風解毒膠囊的批間差異較小、質量較穩定。所生成指紋圖譜的共有模式有22個共有峰,其中指認了(R,S)-告依春、虎杖苷、連翹酯苷A、柴胡皂苷a、柴胡皂苷d、齊墩果酸、熊果酸、連翹苷、大黃素9個共有峰。9個成分含量測定結果顯示,虎杖苷質量分數為8.314~6.058 mg·g-1,標準規定不得少于3.0 mg/粒(5.769 mg·g-1),均符合規定;連翹苷質量分數為1.314~0.856 mg·g-1,標準規定不得少于0.20 mg/粒(0.384 mg·g-1),均符合規定;(R,S)告依春質量分數為0.642~0.427 mg·g-1,柴胡皂苷a質量分數為2.424~1.857 mg·g-1,柴胡皂苷d質量分數為2.364~1.699 mg·g-1,連翹酯苷A質量分數為2.562~1.897 mg·g-1,大黃素質量分數為4.629~2.965 mg·g-1,齊墩果酸質量分數為2.047~1.179 mg·g-1,熊果酸質量分數為2.689~1.286 mg·g-1,不同批次間各組分含量存在一定差異,考慮與不同生產時間各批次間原藥材質量差異有關。
本實驗建立了疏風解毒膠囊UPLC指紋圖譜,并同時測定9個成分的含量,方法穩定簡單,能系統快速地評價疏風解毒膠囊的藥品質量,對全面控制疏風解毒膠囊的質量具有指導意義。
