不同晶體生長活化能對SrZrO3∶Ce發光性能及微觀組織影響
2021-06-19 07:33:14齊鵬遠戴時雨徐曉辰
無機化學學報 2021年6期
馬 瀾 齊鵬遠 馬 雷 戴時雨 徐曉辰 劉 楊
(1沈陽市規劃設計研究院有限公司,沈陽 110004)
(2營口理工學院材料科學與工程學院,營口 115014)
(3沈陽化工大學材料科學與工程學院,沈陽 110142)
堿土SrZrO3∶Ce材料屬鈣鈦礦結構,其高密度(5.42 g·cm-3)、低熱導率、高熱膨脹系數等特點使其在電、磁和發光等方面具有獨特而優開的性能[1-3]。作為稀土發光的基質材料,SrMO3∶Ce(M=Zr、Hf)已被廣泛研究[4-7]。常規濕化學工藝有溶膠-凝膠法、微乳液法、水熱法、共沉淀法等[8-13],其中共沉淀工藝相對簡單,可實現離子均勻摻雜,影響因素易實現可控,大多用反向滴定條件,并且已經得到工業生產的普遍應用。由于不同沉淀劑是合成粉體不可忽視的重要參數,因此,研究粒子合成過程中的熱分析動力學,計算表觀活化能,解決不同沉淀劑制備條件的優選問題至關重要。我們采用反向共沉淀工藝分別用單相和復相沉淀劑制備了SrZrO3∶Ce納米粒子,由熱分解過程的熱重-差熱分析(TG-DTA)曲線,研究合成動力學,利用Doyle-Ozawa積分法和Kissinger微分法計算2種沉淀物在熱分解階段的反應活化能、反應級數、頻率因子等參數,得到不同沉淀物的表觀活化能和晶體生長活化能,討論了不同粒子形貌對樣品發光特性的影響,分析了真空燒結SrZrO3∶Ce陶瓷樣品的顯微組織。
1 實驗部分
原料為硝酸鍶Sr(NO3)2(99.99%)、Ce(NO3)3(99.99%)、氧氯化鋯(ZrOC12·8H2O,99.00%)和硝酸(HNO3)、無水乙醇(C2H6O)、草酸(C2H2O4)、氨水(NH3·H2O)和碳酸氫銨(NH4HCO3)等化學試劑。按照Sr0.997Ce0.003ZrO3化學計量比分別稱取原料Sr(NO3)2、ZrOC12·8H2O、Ce(NO3)3。將 ZrOCl2·8H2O 放入一定量濃 HNO3中,得到 0.15 mol·L-1ZrO(NO3)2溶液;再將Sr(NO3)2溶于上述溶液,配制Zr4+、Sr2+濃度分別為0.15 mol·L-1的母鹽溶液。配制pH=5.5、C2H2O4濃度為0.1 mol·L-1的溶液為單相沉淀劑;配制pH=11.5、NH3·H2O 濃度為 0.2 mol·L-1和 NH4HCO3濃度為 0.3 mol·L-1的混合溶液為復相沉淀劑。分別采用反向滴定法得到前驅體沉淀物,設定蠕動泵滴定速率為2 mL·min-1,反應液體燒杯嵌套到放有冰塊的容器中使反應溫度達到0℃,經磁力攪拌分別獲得2種前驅體沉淀物,濾紙過濾后,再陳化16 h,用C?H?O和去離子水分別洗滌3次,清除雜質離子。然后放入氧化鋁坩堝中在真空干燥箱90℃烘干24 h,前驅體經研磨后再經馬弗爐1 000℃煅燒2 h獲得Ce3+摻雜的物質的量分數為0.3%的SrZrO3樣品。用真空燒結爐(壓力小于10-4Pa)在1 760℃下燒結保溫4 h后降到室溫,得到用復相和單相沉淀劑制備的尺寸30 mm×5 mm×5 mm的陶瓷樣品。
采用D/max-2500PC型X射線衍射儀(XRD)分析不同樣品的物相,CuKα輻射(波長λ=0.154 06 nm),管電壓為40 kV,管電流為200 mA,掃描速率為4(°)·min-1,掃描范圍15°~85°;用HITACHI S-3400N型掃描電鏡(SEM)分析不同樣品形貌及微觀組織,工作電壓為20 kV;采用NET ZSCH STA 449C型差熱失重分析儀(TG-DTA),在氬氣流量120 mL·min-1、溫度變化區298~1 373 K、升溫速率分別為5、10、15、20 K·min-1的條件下,進行前驅體的晶化處理;用Perkin Elmer LS-55型熒光光度計分析不同樣品的發光性能。
2 結果與討論
2.1 不同溫度對物相的影響
圖1a、1b分別為用單相和復相沉淀劑制備的前驅體在不同溫度下煅燒2 h的XRD圖。由圖可知,900℃煅燒時出現SrZrO3∶Ce的晶相特征峰,同時存在少量雜相SrCO3,隨著溫度升高至1 000℃時,雜峰消失,樣品特征峰對應SrZrO3(PDF No.44-0161)的(121)、(202)、(042)、(242)、(323)晶面,屬于立方晶系,表明溫度升高特征峰逐漸增強,半高寬變窄,前驅體結晶逐漸完整,晶化程度得到提高[14]。用2種不同沉淀劑所制備前驅體的XRD圖的特征峰基本相同,說明不同沉淀劑對合成的物相基本沒有影響。

圖1 用單相(a)和復相(b)沉淀劑制備的前驅體在不同煅燒溫度下保溫2 h的XRD圖Fig.1 XRD patterns of the precursor prepared with single phase(a)and multiphase(b)precipitants calcinated at different temperatures for 2 h
2.2 形貌分析
如圖2a、2b分別為用單相和復相沉淀劑制備的前驅體經1 000℃煅燒2 h的SEM圖。前者的粒徑約80 nm,呈棱柱形(圖2a)。后者粒徑約60 nm,為近球形(圖2b)。其形貌及粒徑存在差開的主要原因:單相沉淀劑的pH(5)低于復相沉淀劑的pH(11),這將導致混合溶液飽和析出的前后狀態以及粒子形核和晶核生長速率的不一致。當草酸做沉淀劑時,前驅體是草酸鹽復合物,其在高溫下分解出CO和CO2后直接轉化為SrZrO3∶Ce,由于形核結晶速度較快等因素的影響,生長出棱柱形粒子。當以NH3·H2O和NH4HCO3做復合沉淀劑時,前驅體是碳酸鹽復合物,過量的NH4+吸附在晶核表面,有效控制NH4+離子濃度和分散速率能夠驅動前驅體分子級的形核,有利于近球形晶核的形成且在一定程度上抑制了核的生長[15]。通過控制體系中溶質的相對過飽和度來控制形核速率,并使其晶核形成過程與長大過程分開,就可以形成晶核的同步生長,得到尺寸均勻分布的單分散先驅沉淀物[16-18]。因此,雖然2種形貌的粒子粒徑大小基本均勻,但由于不同沉淀劑與母鹽溶液反應過程中形成的前驅體不同(草酸鹽和碳酸鹽的復合物),在一定程度上使粒子形貌存在明顯的差開[19]。

圖2 用單相(a)和復相(b)沉淀劑制備的前驅體在1 000℃煅燒2 h的SEM圖Fig.2 SEM images of precursors prepared with single phase(a)and multiphase(b)precipitants at 1 000℃for 2 h
2.3 樣品的表觀活化能
圖3分別為用單相和復相沉淀劑制備的前驅體在不同升溫速率下對應的TG-DTA曲線,分別利用Doyle-Ozawa積分法和Kissinge微分法計算樣品各反應階段的表觀活化能。Doyle-Ozawa法計算是指在轉化率α(從失重曲線中質量變化數據算出的)一定時,以lgβ對1/T做圖,由直線的斜率-0.456 7E/R(其中,E為表觀活化能,R為氣體常數,取值8.314 J·K-1·mol-1)計算樣品在分解過程中不同反應階段的表觀活化能,其中β為升溫速率,r為相關系數。表1和表2分別為用單相和復相沉淀劑制備的前驅體在不同升溫速率下,3個主要反應階段所對應的轉化率的溫度。
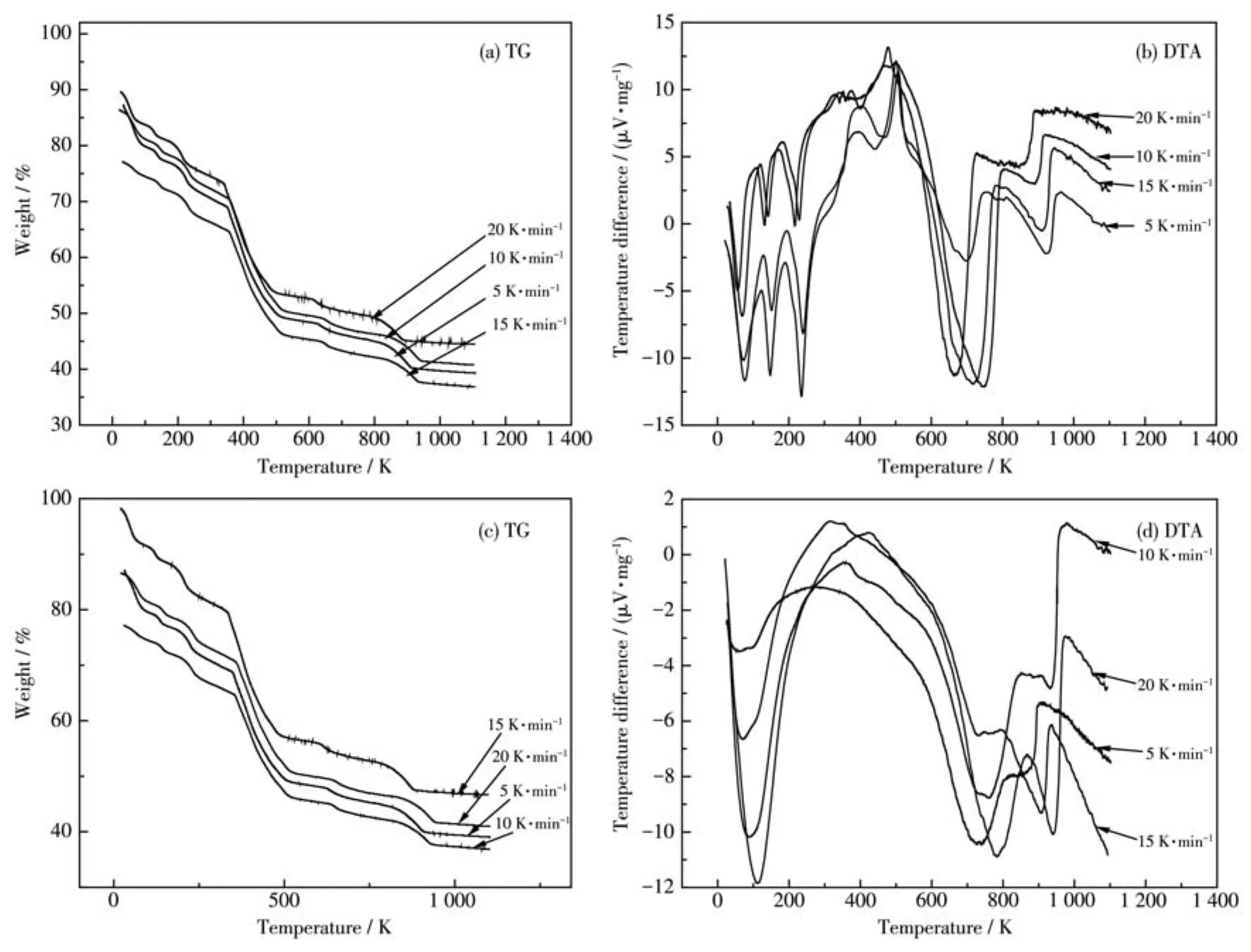
圖3 用單相(a、b)和復相(c、d)沉淀劑制備的前驅體在不同升溫速率下的TG-DTA曲線Fig.3 TG-DTA curves at different heating rates of precursors prepared with single phase(a,b)and multiphase(c,d)precipitants
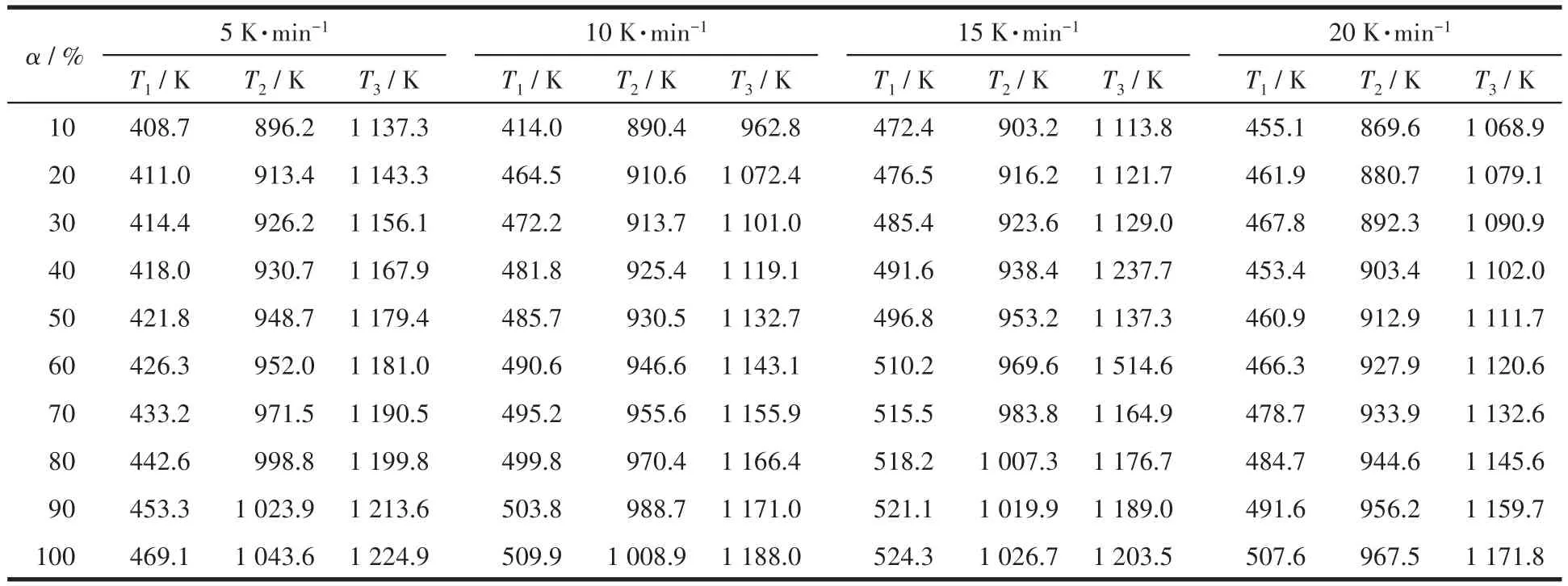
表1 各個反應階段用單相沉淀劑制備的前驅體在不同升溫速率和不同反應轉化率下對應的溫度Table 1 Corresponding temperatures of the precursors prepared with single phase precipitator at different heating rates and different reaction conversion rates in each reaction stage
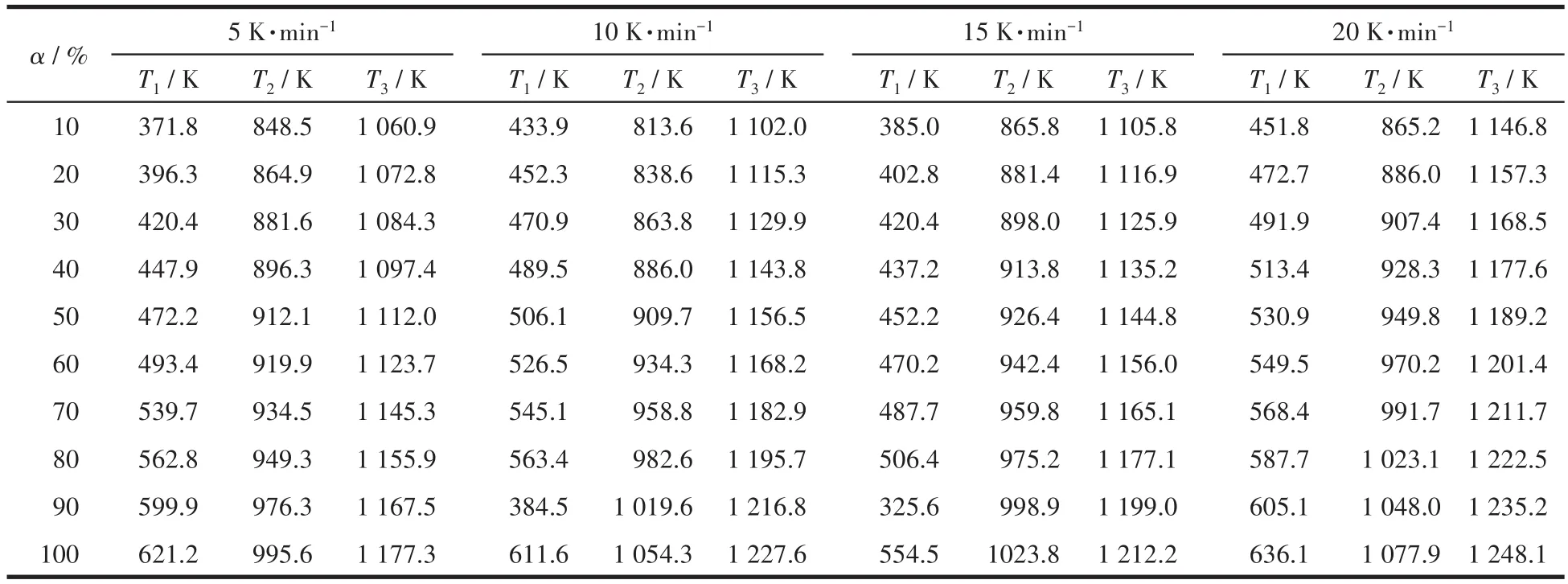
表2 各個反應階段用復相沉淀劑制備的前驅體在不同升溫速率及不同反應轉化率下對應的溫度Table 2 Corresponding temperatures of the precursors prepared with multiphase precipitator at different heating rates and different reaction conversion rates in each reaction stage
圖4和圖5分別為用單相和復相沉淀劑制備的前驅體在各反應階段的lgβ-1/T圖,由圖中各直線斜率-0.456 7E/R計算出不同轉化率時各反應階段的表觀活化能,取其平均值(表3)。從表3看出用單相和復相沉淀劑制備的前驅體表觀活化能分別為95.57、68.06、270.07 kJ·mol-1和 90.98、52.28、233.58 kJ·mol-1,可見后者明顯小于前者,表明復相沉淀劑能降低SrZrO3∶Ce納米粒子合成所需能量,有利于其在較低溫度下的合成。

圖4 Doyle-Ozawa法計算用單相沉淀劑制備的前驅體吸熱峰的lg β-1/T圖Fig.4 lg β-1/T diagrams of the heat absorption peak of the precursor prepared with single phase precipitator calculated by Doyle-Ozawa method
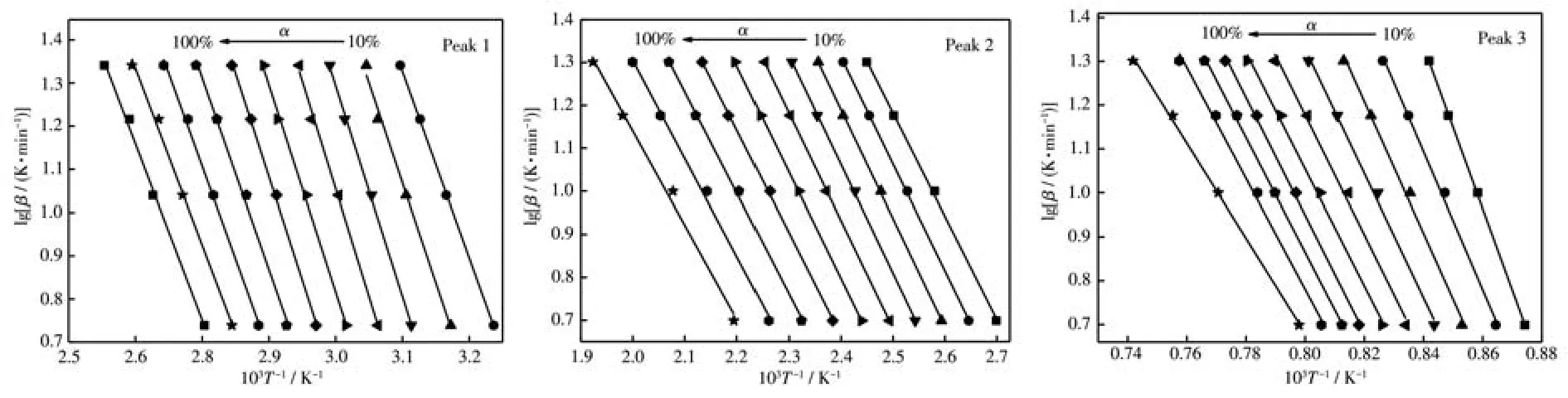
圖5 Doyle-Ozawa法計算用復相沉淀劑制備的前驅體吸熱峰的lg β~1/T圖Fig.5 lg β~1/T diagrams of the heat absorption peak of the precursor prepared with the multiphase precipitant calculated by Doyle-Ozawa method
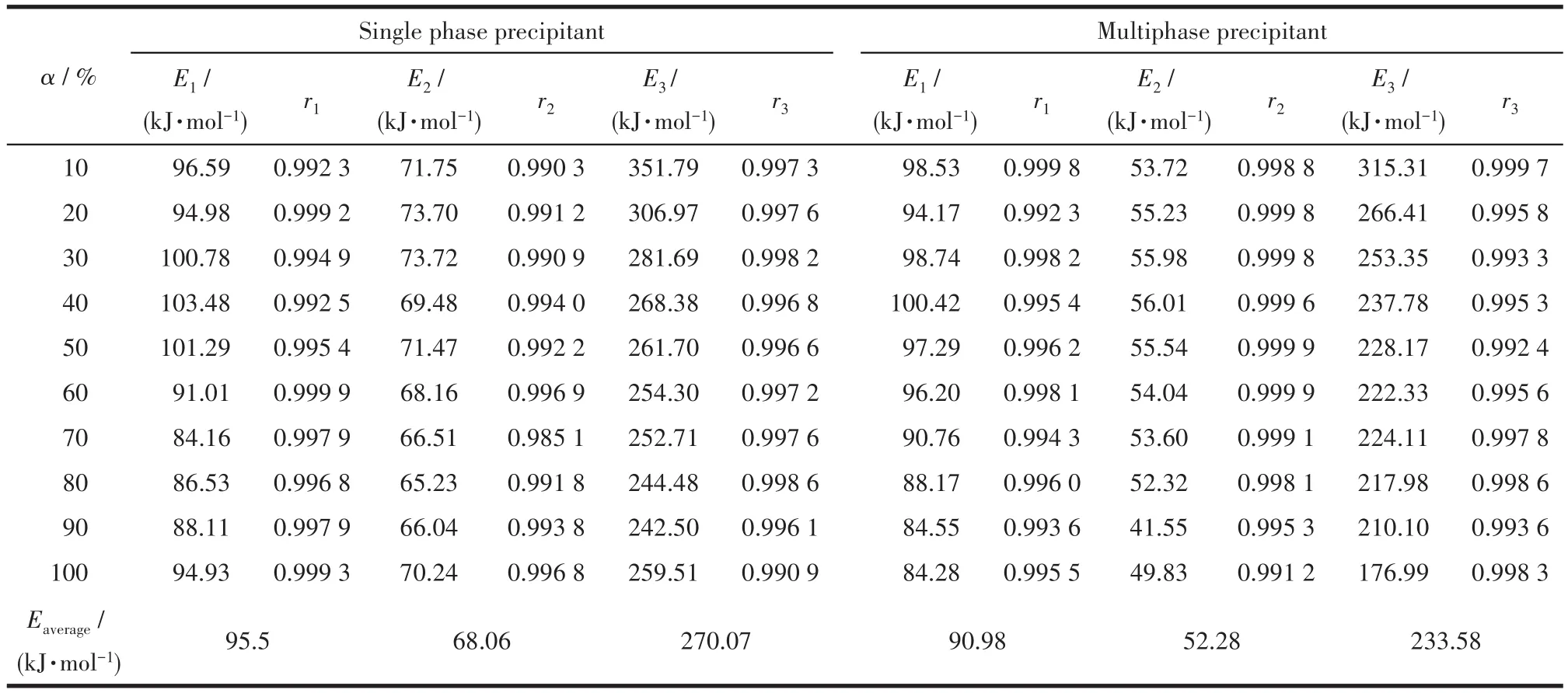
表3 用2種沉淀劑制備的前驅體在不同反應階段中不同轉化率下對應的表觀活化能和相關系數Table 3 Apparent activation energy and correlation coefficient corresponding to different conversion rates in each reaction stage of the precursors prepared with two precipitants
Kissinger法的公式表明 ln(β/Tm2)與 1/Tm呈直線關系,其中Tm為峰值溫度,因此可通過以ln(β/Tm2)對1/Tm作圖,根據斜率-E/R計算出反應活化能,如圖6所示。表4為由圖6中的直線斜率計算得到的表觀活化能及相關系數的平均值,可見前者數值明顯較大,這與Doyle-Ozawa法計算所得的數據規律相同。
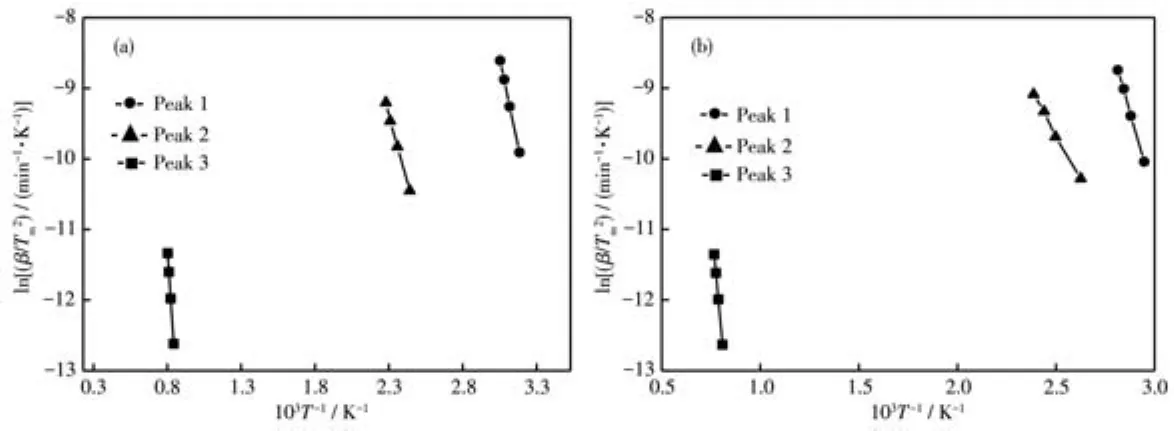
圖6 用單相和復相沉淀劑制備的前驅體吸熱峰在不同升溫速率下的ln(β/Tm2)-1/Tm圖Fig.6 ln(β/Tm2)-1/Tm diagrams of heat absorption peaks of the precursor prepared with single phase(a)and multiphase(b)precipitants at different heating rates
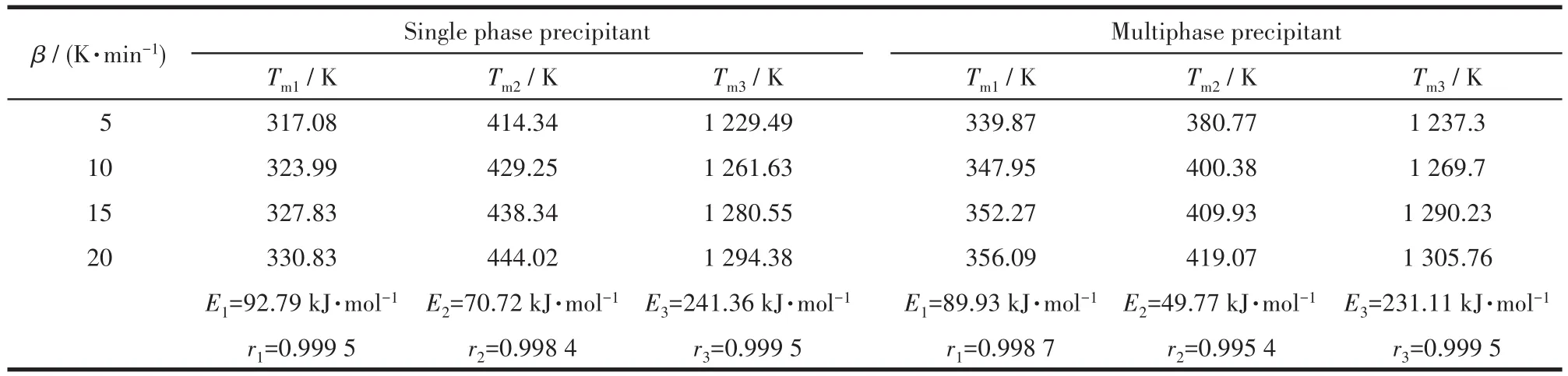
表4 用單相和復相沉淀劑制備的前驅體在不同升溫速率下的峰值溫度、表觀活化能及相關系數Table 4 Peak temperature,apparent activation energies and correlation coefficient of the precursors prepared with single phase and multiphase precipitant at different heating rates
由表5可知,用單相和復相沉淀劑制備的前驅體在不同反應階段的平均表觀活化能分別為94.18、69.39、255.72 kJ·mol-1和 90.46、51.03、232.35 kJ·mol-1。這表明各個階段的表觀活化能越大,其反應越困難,延長保溫時間可以提供更多能量使反應進行完全。復相樣品的表觀活化能明顯小于單相樣品,分析表明這是由于單相和復相沉淀劑使得反應物前驅體的形核粒度不同,顆粒細小的反應物能夠增大摩爾表面能,減小反應活化能。加入復相沉淀劑時的成核機制有利于生成粒度細小且分布均勻的前驅沉淀物(圖2b),因此,復相沉淀劑是制備SrZrO3∶Ce粒子首選工藝方法。
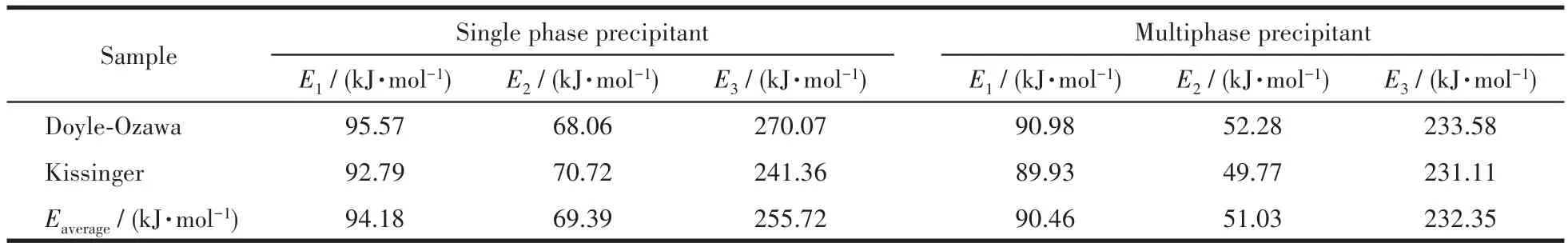
表5 Doyle-Ozawa法和Kissinger法計算用單相和復相沉淀劑制備的前驅體在各反應階段的表觀活化能Table 5 Apparent activation energies of the precursors prepared with single phase and multiphase precipitant calculated by Doyle Ozawa method and Kissinger method
2.4 樣品的晶體生長活化能
采用XRD圖(圖1)中的特征峰數據繪制的lnD~1/T曲線,如圖7所示。由二者的直線斜率-E/R可知,復相沉淀劑對應SrZrO3∶Ce粒子的線性斜率明顯低于單相的斜率,計算出其晶體生長活化能分別為E復相=22.53 kJ·mol-1和E單相=27.97 kJ·mol-1。
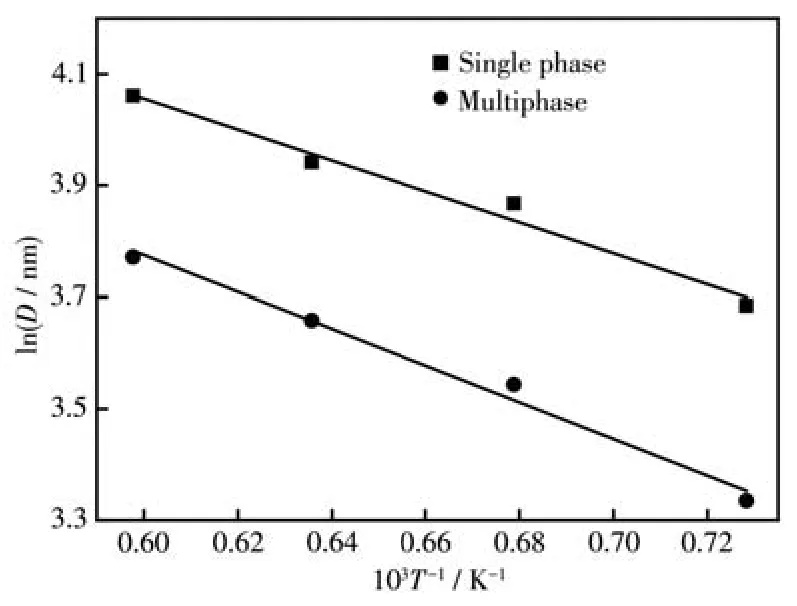
圖7 SrZrO3∶Ce粒子的ln D和1/T關系曲線Fig.7 Relationship between ln D and 1/T for SrZrO3∶Ce particles
2.5 樣品的發光性能
圖8為用不同沉淀劑制備的SrZrO3∶Ce粉體粒子的激發光譜和發射光譜。由圖8a可知,二者在225~300 nm處均存在較寬的激發光譜帶,峰值分別在約255 nm波長處且用復相沉淀劑所制備樣品的相對峰值強于單相,對應于Ce3+離子4f能級向5d能級的躍遷吸收[20]。由于Ce3+在4f能級只有一個電子,自旋軌道耦合使4f能級分解為2個能級2F5/2和2F7/2,Ce3+離子進入Sr2+離子的晶格位置,基質晶格缺陷處晶場的強度為Ce3+離子激發能的吸收提供了傳遞能量的環境,4f能級由于受到外層5s25p6電子的屏蔽,仍然保持自由離子時的LS耦合能級的特征[21-22]。5d能級在受晶場的作用以及在低對稱條件下,實現了Ce3+電子能級的躍遷,從而出現激發光譜峰。除直接激發的傳遞能量外,還有從基質晶場到Ce離子的能量傳遞,造成譜峰寬化。
在247 nm波長激發下,2種樣品均存在較寬的發光譜帶(425~495 nm波段),用復相和單相沉淀劑制備的粉體樣品的峰值在455 nm處,前者相對強度高于后者(圖 8b),對應 Ce3+的 5d→2F5/2和 5d→2F7/2發光躍遷[16]。分析表明用復相沉淀劑制備的SrZrO3∶Ce納米粒子的發光性能明顯強于單相沉淀劑的。盡管制備的SrZrO3∶Ce粒子粒度細小、分布均勻,但前者為近球形的形貌,后者為棱柱形的形貌,其比表面積、粒子大小等特性是前者優于后者,另外晶體生長活化能大小可能與發光強度存在關聯性。復相樣品活化能(22.53 kJ·mol-1)明顯低于單相(27.97 kJ·mol-1),粒子活化能越小其相對發光強度越大,與圖8相對應。可認為不同形貌樣品的內外表面附近的Ce3+的共價性和感受到的晶場強度與體相中的Ce3+的作用不同,顯然2種樣品Ce3+的光躍遷所需能量的微小差開以及存在結構缺陷也可造成譜峰加寬和峰值強弱的不同。不同粉體顆粒的形貌、尺寸、組成和表面狀態等都會影響其發光性能,分散性越好,發光性能越強[11-12],與上述分析中前者優于后者的結果具有一致性。

圖8 用不同沉淀劑所制備的SrZrO3∶Ce納米粒子的激發光譜(a)和發射光譜(b)Fig.8 Excitation spectra(a)and emission spectra(b)of SrZrO3∶Ce nanopaticles prepared with different precipitants
2.6 不同陶瓷樣品的微觀組織
圖9是經1 760℃真空燒結并保溫4 h條件后用復相和單相沉淀劑制備的陶瓷樣品的SEM圖。由于兩者的粉體形貌不同,經相同條件燒結后前者的晶粒尺寸大小不一(圖9a),這可能源于共沉淀工藝所制備的粉體粒徑存在呈正態分布的尺寸差,以致在真空燒結的多因素影響下導致大晶粒吞噬小晶粒以及晶粒生長取向、擴散形式的不確定性,使晶粒大小不均勻。相對用復相沉淀劑制備的燒結樣品而言,單相對應的晶粒存在較多開常長大的現象,其原因是其具有更高的活化能,不利于燒結過程的傳質擴散。可見粒子活化能對粉體材料的燒結致密化起到重要作用,活化能越小越有利于提高燒結驅動力,從而增強顆粒系統的高能態向低能態的轉變能力,粉體粒子活化能直接影響到晶粒的擴散和物質遷移能力,進而使微觀組織相對均勻,并促進了燒結致密化[23-25]。

圖9 經1 760℃真空燒結后用復相(a)和單相(b)沉淀劑制備的陶瓷樣品的微觀組織(表面熱蝕)Fig.9 Microstructure of ceramics prepared with multiphase(a)and single phase(b)precipitants after sintered at 1 760℃in vacuum(surface thermal erosion)
3 結論
(1)采用反向共沉淀法以草酸為單相沉淀劑,氨水和碳酸氫銨為復相沉淀劑制備了SrZrO3∶Ce納米粉體。當滴定速率為2 mL·min-1,體系溫度為0℃,陳化時間為16 h,經1 000℃煅燒2 h后分別獲得分散良好的棱柱形和近球形粒子,粒徑約80和60 nm。
(2)用單相和復相沉淀劑制備的前驅體的表觀活化能分別是94.18、69.39、255.72 kJ·mol-1和90.46、51.03、232.35 kJ·mol-1。晶體生長活化能分別為E單相=27.97 kJ·mol-1和E復相=22.53 kJ·mol-1,后者的表觀活化能和晶體生長活化能分別小于前者,表明復相沉淀劑降低了粒子的合成能量,提高了SrZrO3∶Ce粒子的燒結活性。
(3)用不同沉淀劑制備的SrZrO3∶Ce納米粒子在441 nm的激發波長作用下,最大峰值約在255 nm處出現;在247 nm的發射波長作用下,發光強度在約255 nm處存在最大峰值。激發和發射光譜均為較寬帶譜,激發光譜分別在247和441 nm處存在最大峰值。發光機制是摻雜的Ce3+離子受激發產生對應5d→2F5/2和5d→2F7/2發光能級帶的躍遷。可見,用復相沉淀劑制備的SrZrO3∶Ce納米粒子的發光強度明顯優于單相沉淀劑對應的樣品。
(4)在1 760℃真空燒結并保溫4 h后用復相和單相沉淀劑制備的陶瓷樣品的顯微組織存在著差開。前者晶粒不均勻性是晶粒生長取向、擴散形式的不確定性引起的。后者部分晶粒開常長大是由于較高活化能的存在不利于燒結過程的傳質擴散,原因是粉體粒子活化能對燒結致密化起到重要作用,活化能越小越有利于提高燒結驅動力,促進顆粒體積、表面、晶界擴散及物質遷移能力,使微觀組織均勻,實現了燒結致密化。

- 無機化學學報的其它文章
- Synthesis and Characterization of Palladium Nanoparticles with High Proportion of Exposed(111)Facet for Hydrogenation Performance
- Syntheses,Crystal Structures,Luminescence and Catalytic Activity of Manganese(Ⅱ)and Cadmium(Ⅱ)Coordination Polymers Based on 2,3-Dihydroxy-terephthalic Acid
- Scale-Up Strategy to Develop Highly-Effective Co-N-C@KB Composites as Sulfur Host for Lithium-Sulfur Battery
- Self-Assembled Zn2+,Co2+ and Ni2+ Complexes Based on Coumarin Schiff Base Ligands:Synthesis,Crystal Structure and Spectral Properties
- Synthesis,Characterization,and X-ray Crystal Structure Analysis of Cu(Ⅰ)/Cu(Ⅱ)Complexes of Phenanthridine and Triphenylphosphine
- 添加叔丁醇鉀對Mg(NH2)2-2LiH體系儲氫性能的影響