
改良QuEChERS-LC-MS/MS測定藥食同源食品苯基吡唑類農藥殘留
2021-06-28 14:22:26陳婷閆君張文彭濤吳福祥
食品工業 2021年6期
關鍵詞:方法
陳婷,閆君,張文,彭濤,吳福祥
蘭州市食品藥品檢驗檢測研究所(蘭州 730050)
中國有著悠久的飲食文化及中醫藥文化,藥食同源食品由于其兼備了獨特的營養與保健功效,近年來受到人們的廣泛青睞[1-2]。2019年衛建委公布了按照傳統既是食品又是中藥材的物質目錄,從原有101個品種新增至107種[3],藥食同源產品的研發、生產、應用等活動也逐漸成為當今熱點話題[4]。
藥食同源物質以可食性植物為主,主要通過栽培種植獲得。為滿足市場的需求,在栽培過程中常盲目濫用禁限用農藥防治病、蟲害,致使藥食同源類植物受到農藥污染日益嚴重。農藥殘留不僅影響藥食同源類產品在國際市場的貿易,更重要的是對人類健康造成威脅[5-6]。苯基吡唑類農藥可以有效地殺死各種蟲類,曾被廣泛地運用到農作物中,其中最常見的苯基吡唑類是丁蟲腈、氟蟲腈及其代謝物,人體攝入后會嚴重影響神經系統并且會損傷肝臟、甲狀腺等,且產生較為明顯的富集現象[7-8]。我國要求自2009年7月1日起,除衛生用、部分旱田種子包衣劑外,停止銷售和使用含有氟蟲腈成分的農藥制劑,歐盟于2013年起對氟蟲腈實行限制使用,禁止氟蟲腈在食品產業畜禽養殖中使用,因此建立苯基吡唑類農藥快速檢測方法尤為重要。目前,針對此類農藥的檢驗方法較少,2015版《中國藥典》通則2341《農藥殘留測定法》中只針對氟蟲腈一種化合物進行檢測,且分析時間較長[9]。國標方法只涉及雞蛋基質中氟蟲腈及其代謝物殘留量的檢測方法,此外其他行業標準采用固相萃取法進行檢驗,其操作過程復雜,耗時長,回收率不穩定。近年來QuEChERS方法在農藥殘留檢測中應用較廣,常用的凈化材料主要有十八烷基鍵合硅膠(C18)、乙二胺-N-丙基硅烷(PSA)、石墨化炭黑(GCB)、多壁碳納米管(MWCNTs)等[10]。石墨化多壁碳納米管(Graphitized multi-wall carbon nanotubes,GMWCNTs)是一種新型納米材料,由于具有納米級別的中空管結構和較大的比表面積,表現出獨特的吸附能力,經高溫石墨化處理可以有效減小納米管粉體內的非晶碳,進而消除管狀結構內的缺陷[11-12]。目前,利用GMWCNTs作為凈化材料在藥食同源食品農藥殘留分析領域鮮有報道。因此,試驗采用新興凈化材料GMWCNTs并結合超高效液相色譜質譜聯用技術建立藥食同源食品中苯基吡唑類農藥的快速檢測方法,為相關監管部門的日常抽檢和風險監測提供技術支撐。
1 試驗部分
1.1 儀器與試劑
Waters Iclass/Xevo TQD高效液相色譜質譜聯用儀(配有電噴霧離子源、三重四極桿線性離子阱質量分析器及MassLynx 4.1工作站,美國Waters公司);SiO-6512 QuEChERS全自動樣品制備系統(配有渦流振動離心機,12位均質離心轉子,均質分離工作站,北京ABILITY公司);EVA50A氮吹儀(北京普立泰科儀器有限公司,中國);Milli-Q超純水機(美國Millipore公司);Centrifuge 5810R高速離心機(德國Eppendorf公司);VORTEX-5渦旋混勻器(海門市其林貝爾儀器制造有限公司);ME204/02電子天平(美國梅特勒公司)。
乙腈、丙酮、甲醇、甲酸(色譜純,德國Merck公司);試驗用水(美國Millipore公司);提取劑包(4.0 g無水硫酸鎂,1.0 g氯化鈉,1.0 g檸檬酸鈉,0.50 g檸檬酸二鈉,均為分析純)、N-丙基乙二胺(PSA)、石墨化多壁碳納米管(GMWCNTs)、聚丙烯整合管、ZrO2珠包(北京Ability公司);石墨化氨基固相萃取柱(美國安捷倫公司);農藥標準品(純度≥95%,德國Dr Ehrenstorfer公司)。
1.2 試驗條件
1.2.1 標準溶液的配制
1.2.1.1 標準儲備溶液
分別準確稱取10 mg(精確至0.01 mg)丁蟲腈、氟蟲腈、氟甲腈、氟蟲腈砜和氟蟲腈亞砜標準品于10 mL容量瓶中,用丙酮溶解并定容至刻度,于-18 ℃避光保存。用甲醇稀釋標準儲備液,配制成一定濃度的混合標準工作液,于4 ℃避光保存,備用。
1.2.1.2 空白基質溶液的制備
取空白樣品,按1.2.3小節處理,過0.22 μm微孔濾膜后,于4 ℃保存備用。
1.2.2 試樣制備
樣品干燥后粉碎處理,過50目篩(篩孔0.300 mm),在常溫下保存。
1.2.3 試驗方法
準確稱取2 g試樣,精確至0.01 g,分別至整合管外管,依次加入5 mL超純水、ZrO2珠包(24粒)、10.0 mL乙腈、6.5 g QuEChERS鹽包。將裝有凈化劑的內管(凈化劑為0.9 g MgSO4、0.2 g PSA、0.18 g GMWCNTs)放入整合管并擰緊,放入SiO-6512主機中。運行方法:第Ⅰ步,1 100 r/min震動5 min;第Ⅱ步,以4 000 r/min離心10 min;第Ⅲ步,以1 100 r/min振動5 min;第Ⅳ步,以4 000 r/min離心5 min。程序結束后取出整合管,準確吸取1.00 mL上清液過0.22 μm微孔濾膜,待LC-MS/MS測定。
1.2.4 儀器方法
1.2.4.1 液相色譜條件
色譜柱:ACQUITY UPLC BEH C18(1.7 μm,2.1 mm I.D.×100 mm);柱溫35 ℃;進樣量1.0 μL;流速0.3 mL/min;流動相:乙腈(A)-0.1%甲酸水(B);梯度洗脫:0~0.5 min,5% A;0.5~8.5 min,5%~95% A;8.5~10.0 min,95% A;10.0~12.0 min,5% A。
1.2.4.2 質譜條件
電離方式:電噴霧離子源(ESI-);掃描方式:多重反應監測(MRM);去溶氣溫度500 ℃;去溶氣流速900 L/Hr;毛細管電壓3.00 kV;錐孔電壓45 V;離子源溫度150 ℃;碰撞氣,高純氬氣(99.999%)。各農藥質譜參數見表1。
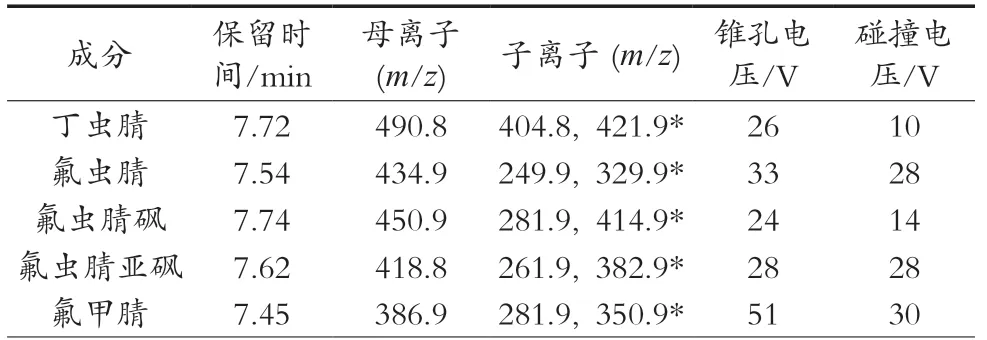
表1 苯基吡唑類農藥的保留時間及質譜參數
2 結果與討論
2.1 凈化劑種類及用量的優化
藥食同源食品基質較為復雜,存在多糖、有機酸、脂肪酸、色素等干擾物,試驗選擇MgSO4、PSA、GMWCNTs作為凈化材料,通過對比不同凈化劑含量、組合對目標化合物回收率的影響,得出最佳的凈化劑組合。
2.1.1 無水硫酸鎂使用量的優化
MgSO4可以有效降低有機相中的水分,有利于GMWCNTs與樣品雜質結合,提高凈化效率。試驗使用2.0 g肉桂空白樣品,通過加標回收優化無水硫酸鎂的使用量。凈化劑由200 mg PSA、180 mg GMWCNTs及不同量的MgSO4(800,900,1 000和1 100 mg)組成。如圖1所示,隨著MgSO4用量的增加,各農藥的回收率逐漸增高,當MgSO4用量達到900 mg時,除氟蟲腈砜以外的化合物回收率均變化不大。為了達到較好的脫水效果且考慮試驗成本,選擇無水硫酸鎂的用量900 mg。
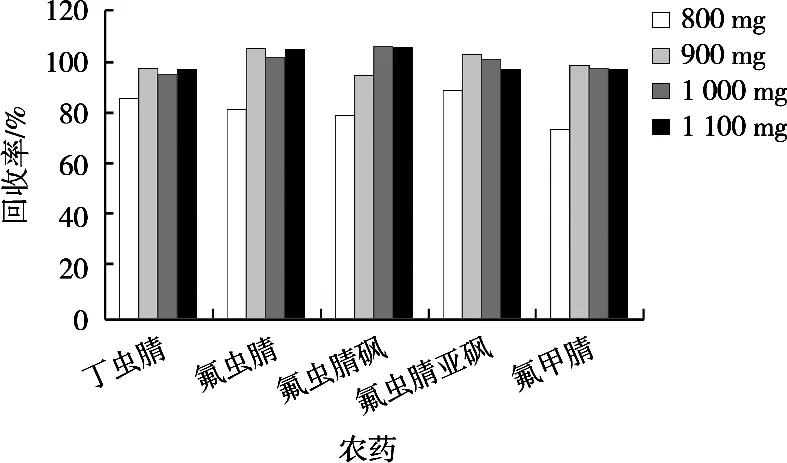
圖1 不同無水硫酸鎂用量對肉桂中農藥回收率的影響
2.1.2 PSA使用量的優化
將2.0 g肉桂空白樣品按1.2.3小節處理,通過加標回收優化PSA使用量。凈化劑由900 mg MgSO4、180 mg GMWCNTs及不同量的PSA(100,150,200和250 mg)組成,結果如圖2所示。當PSA含量較低時,各化合物回收率較低;隨著PSA含量的增加,化合物的回收率逐漸增加;當PSA含量為200 mg時,回收率趨于穩定。因此,試驗選擇200 mg PSA作為凈化最佳用量。
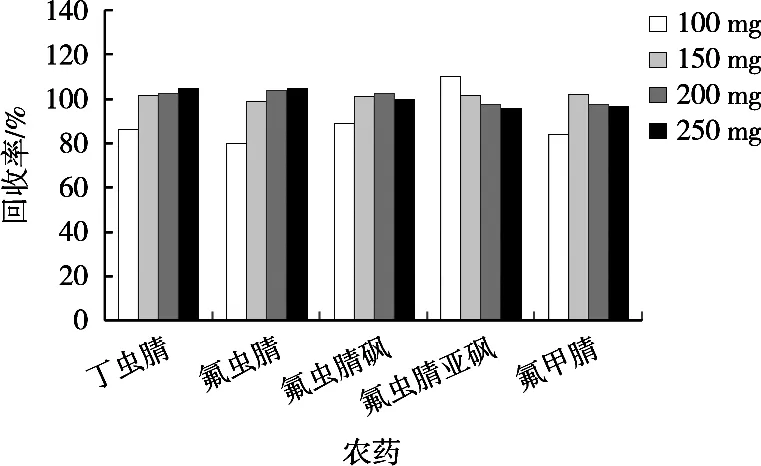
圖2 不同PSA用量對肉桂中農藥回收率的影響
2.1.3 GMWCNTs使用量的優化
將2.0 g肉桂空白樣品,按1.2.3小節處理,通過加標回收優化GMWCNTs使用量。凈化劑由900 mg MgSO4、200 mg PSA及不同量的GMWCNTs(60,100,140,180和220 mg)組成,結果如圖3所示。當GMWCNTs用量為60 mg時,丁蟲腈、氟蟲腈、氟蟲腈砜回收率均小于80%;當用量為180 mg時,各化合物回收率均接近100%。因此,試驗選擇180 mg GMWCNTs作為凈化最佳用量。
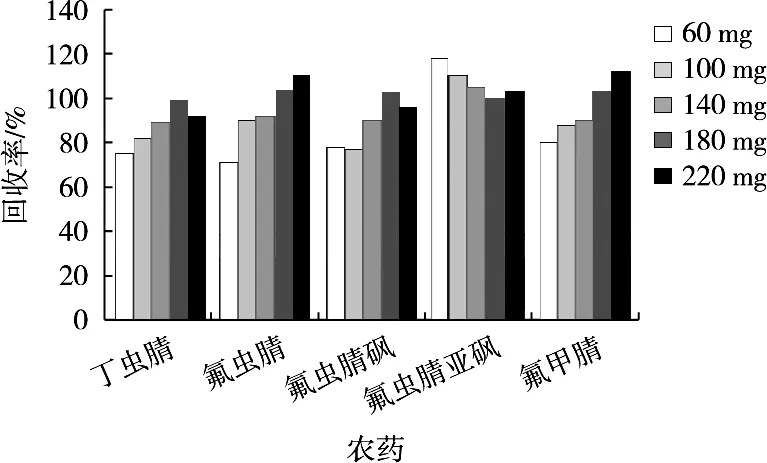
圖3 不同GMWCNTs用量對肉桂中農藥回收率的影響
2.2 與傳統固相萃取方法的比較
為了驗證改良QuEChERS方法的凈化效果,將其與傳統固相萃取方法(采用石墨化氨基固相萃取柱凈化)進行比較。向肉桂空白樣品中添加50.0 μg/kg濃度水平的混合標準溶液,對比兩種方法的凈化效果。圖4(a)和(b)分別顯示了未經凈化處理的肉桂樣品MRM色譜圖與傳統的固相萃取法處理的樣品溶液色譜圖。通過與改良QuEChERS方法凈化后的肉桂樣品色譜圖(圖4c)比較可以看到干擾峰的背景明顯降低,說明該方法對于樣品中含有的多糖、有機酸、醛類、酯類等極性和弱極性物質均能達到良好的凈化效果。
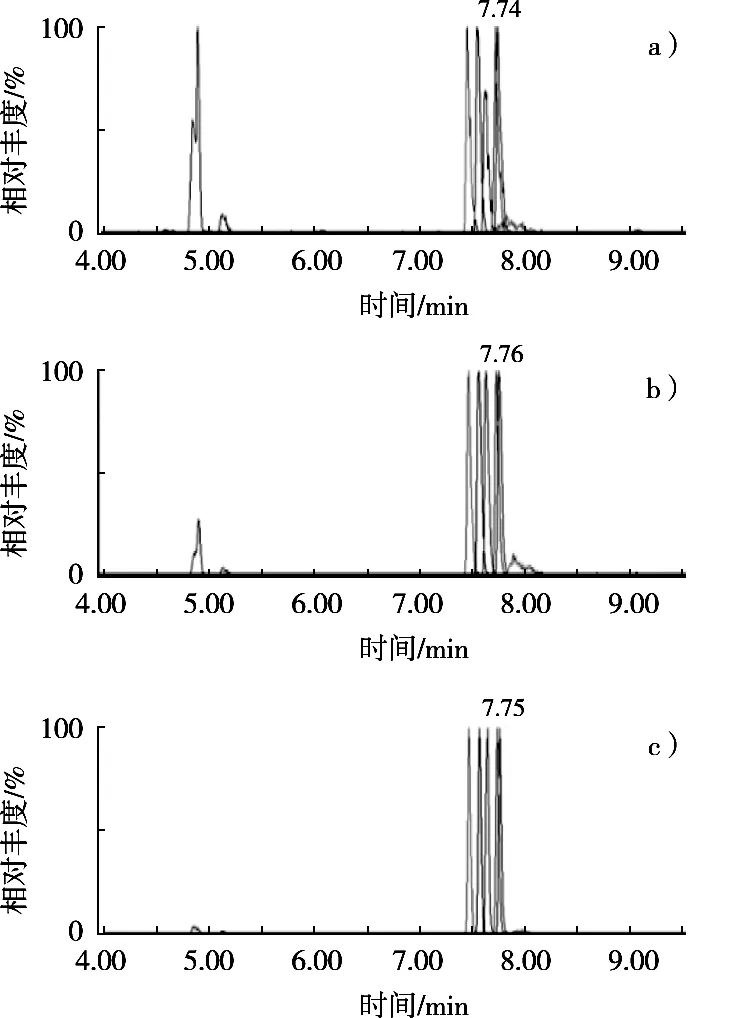
圖4 采用不同凈化方式處理后的各農藥總離子流色譜圖
2.3 基質效應
在質譜法檢測過程中,樣品中的基質成分對目標化合物的離子化有一定的增強或抑制作用,進而影響測定結果的精密度和準確度。試驗通過比較肉桂、金銀花、山藥、葛根空白基質與乙腈配制的混合標準溶液的斜率,評價基質效應。公式:ME=mmatrix/msolvent×100%,其中ME為基質效應,mmatrix、msolvent分別為基質標準曲線和溶劑標準曲線的斜率,比值越接近1,則基質效應越小。表2顯示:不同化合物在不同基質中存在不同的基質效應,不同的樣品基質中肉桂基質效應較大,5種化合物中氟蟲腈基質效應最為顯著。因此,試驗采用相近空白基質配制標準曲線對樣品進行校正。
2.4 方法的線性范圍及檢出限
試驗利用優化的分析方法,分別用陰性樣品空白基質配制不同濃度的混合標準溶液,經UPLC-MS/MS檢測,以化合物質量濃度為橫坐標,定量離子對峰面積為縱坐標,繪制標準曲線。向陰性樣品中添加目標化合物,以樣品峰響應值為3倍和10倍噪音的添加濃度作為方法的檢出限(LOD)和定量限(LOQ)。結果顯示:苯基吡唑類化合物的色譜峰面積(y)與其質量濃度(x,ng/mL)在各自的線性范圍內具有良好的線性關系,線性相關系數均大于0.99。方法的檢出限在2.00~4.38 μg/kg之間,定量限在5.99~13.15 μg/kg之間,結果見表2。
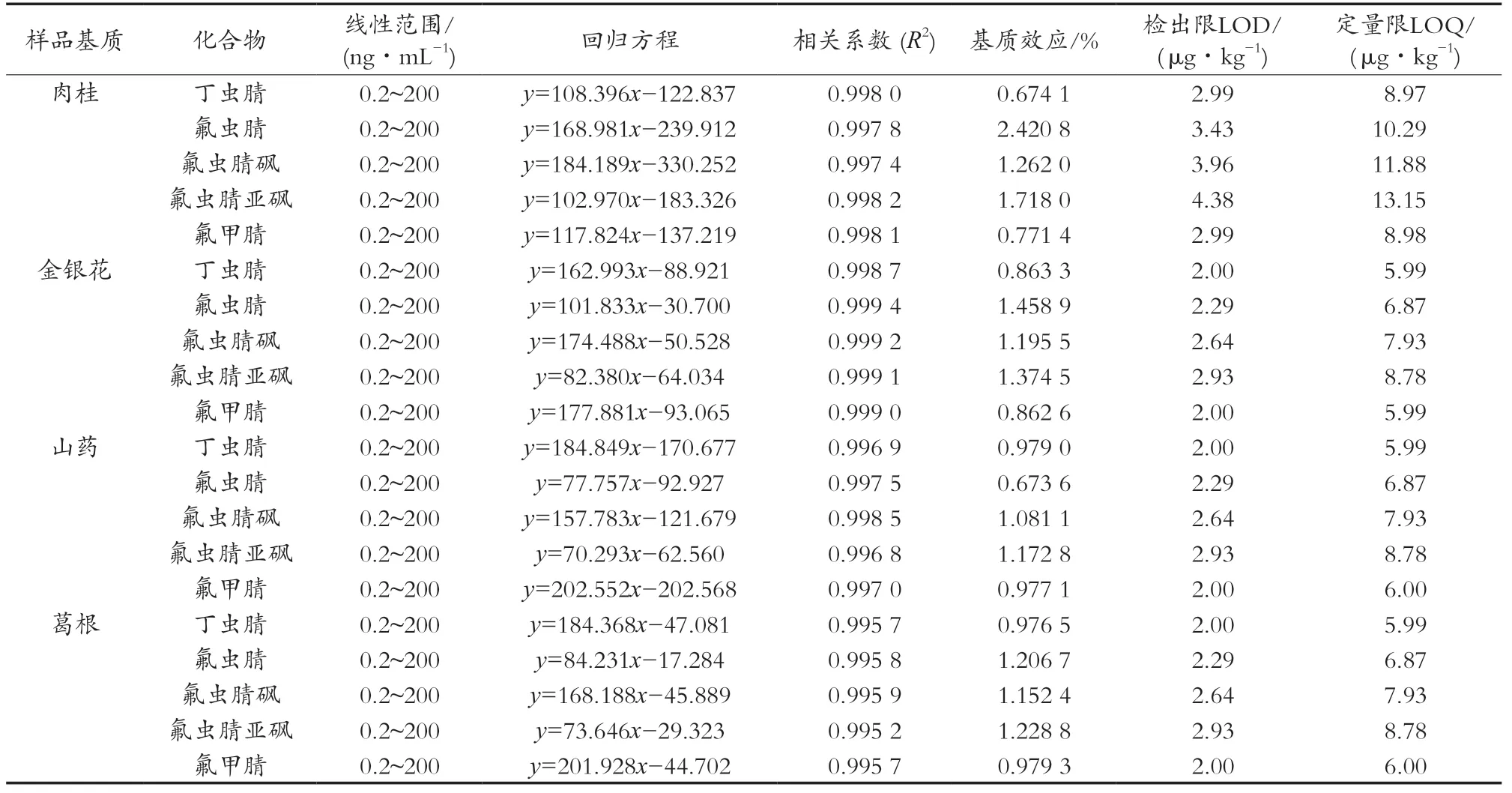
表2 線性范圍、線性方程、相關系數、基質效應及檢出限、定量限
2.5 方法的回收率及精密度
選用陰性樣品肉桂、金銀花、山藥、葛根,分別向其中添加20.0,40.0和80.0 μg/kg三個濃度水平的混合標準溶液,每個添加水平做5次平行,采用基質匹配標曲進行定量分析,考察方法的回收率和精密度。由表3可知,苯基吡唑類農藥的加標回收率在68.8%~128.1%之間,相對標準偏差(δRSD)<10.3%。

表3 不同基質中各農藥的回收率及相對標準偏差(n=5)
2.6 實際樣品檢測
采用試驗建立的分析方法對市售的金銀花、枸杞、葛根、肉桂、山藥、菊花6種藥食同源食品的50批次樣品進行了檢測,其中1批次金銀花中檢出0.018 mg/kg氟蟲腈,其余樣品均未檢出。
3 結論
采用石墨化多壁碳納米管作為前處理凈化劑,并結合UPLC-MS/MS建立了針對藥食同源性食品中苯基吡唑類農藥的改良QuEChERS檢測方法。該方法與傳統的固相萃取法相比,凈化效果良好,有效降低了藥食同源食品中色素、多糖等大分子物質的干擾,同時使用全自動QuEChERS樣品制備系統,避免了前處理過程中人為操作帶來的誤差,進一步簡化了試驗過程。該方法操作簡便有效、準確可靠,可用作藥食同源食品中苯基吡唑類農藥殘留的參考檢測方法。
猜你喜歡
中老年保健(2021年9期)2021-08-24 03:52:04
河北畫報(2021年2期)2021-05-25 02:07:46
中學生數理化(高中版.高考理化)(2020年2期)2020-04-21 05:33:04
兒童繪本(2020年5期)2020-04-07 17:46:30
兒童故事畫報(2019年5期)2019-05-26 14:26:14
意林原創版(2016年10期)2016-11-25 10:28:30
Coco薇(2016年2期)2016-03-22 02:42:52
山東青年(2016年1期)2016-02-28 14:25:23
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年7期)2015-08-11 15:03:12
