自蔓延燃燒法制備Pd0.01CuxCe(1-x)/2Zr(1-x)/2Oy催化劑催化NO制NH3反應的性能研究
2021-06-30 01:10:32閆瑞寧王一男趙旭騰劉旻劉興胡琨石福祿王濤林赫黃震
車用發動機 2021年3期
關鍵詞:催化劑
閆瑞寧,王一男,趙旭騰,劉旻,劉興,胡琨,石福祿,王濤,林赫,黃震
(1.上海交通大學新能源動力研究所教育部重點實驗室,上海 200240;2.上汽汽車乘用車公司技術中心,上海 201804;3.北京市機動車排放管理中心,北京 100176)
與傳統的化學計量發動機相比,稀薄燃燒發動機不僅具有更好的燃油經濟性,而且二氧化碳排放也更低[1]。然而,如何有效地減少稀薄燃燒廢氣中的氮氧化物(NOx)仍然是汽車工業面臨的挑戰。NOx作為主要的大氣污染物,會導致酸雨和城市煙霧的形成,對自然和人類健康造成災難性的影響[2]。因此,最新發布的排放法規如EURO 6[3]和China 6a[4]對NOx排放有嚴格的限制。三效催化劑(TWC)在當量空燃比(λ=1±0.01)下對化學計量發動機排氣中NOx,CO和HC的同時控制非常有效,但在稀薄燃燒條件下性能較差。當λ大于1.2時,由于TWC的衰減效應,NOx排放急劇增加[5]。因此,需要采用新的后處理系統來滿足嚴格排放法規的要求。
通用電氣公司(General Electric Company)首次提出了這種被稱為“被動式”或“無尿素”SCR的新技術[6]。在傳統TWC的基礎上,增加了SCR脫硝,在周期性富燃運行過程中,由TWC供給NH3并將其儲存在SCR催化劑上。而在貧燃運行期間,通過SCR上儲存的NH3降低了NOx。這種所謂的TWC+PSCR法消除了傳統的SCR尿素罐,降低了成本。
具體地說,在富燃過程中,在TWC催化劑上NO與H2及HC生成NH3儲存在下游SCR中;儲存的NH3在稀燃時與NOx發生SCR反應產生N2[7]。被動SCR法的關鍵反應是生成NH3[8]。然而,對TWC催化劑上NH3生成的研究較少。事實上,NH3一直被視為環境的副產品甚至污染物,在TWC反應過程中肯定會產生NH3,并從車上逃逸,這一點已經得到了后續反應器篩選、發動機底盤測功機研究和風洞調查等試驗的證實[9]。Heeb等[10]進一步論證了NH3作為二次污染物應納入今后的法規中。相反,對于被動式SCR脫硝方法,其主要目標是通過TWC生產NH3并輸送到下游SCR催化劑,此反應中NH3是有益的中間產物,用于后續NOx的去除。
自蔓延燃燒合成法由于其能效高、反應時間短、產率高、凈化能力強,在金屬氧化物催化劑領域中受到廣泛的關注[11]。因此,本研究采用自蔓延燃燒合成法對催化劑進行制備,得到純凈及高產率的催化劑。采用Pd/Ce0.5Zr0.5Ox催化劑模擬TWC催化劑的主要成分,并摻雜Cu進行改性,從而提高NOx轉化為NH3的催化能力。
1 實驗
1.1 催化劑制備
采用自蔓延燃燒合成法[12]制備Pd0.01CuxCe(1-x)/2Zr(1-x)/2Oy型貴金屬催化劑。首先按照不同的Pd,Cu,Ce,Zr配比制作前驅液,將Pd(NO3)2·xH2O,Cu(NO3)2·9H2O,Ce(NO3)3·6H2O,Zr(NO3)4·5H2O粉末溶于20 ml去離子水中,并按照一定的化學計量比(甘氨酸與硝酸根離子比例為5∶9)加入甘氨酸(C2H5NO2)作為助燃劑。將混合溶液在60 ℃下充分攪拌1 h后轉移至坩堝內,在馬弗爐中迅速升溫到600 ℃并持續焙燒4 h,最終將坩堝內燃燒所得的粉末經過壓片、研磨等處理后篩分至40~80目大小的顆粒備用。按照不同的Pd,Cu,Ce,Zr配比,將催化劑標記為Pd0.01CuxCe(1-x)/2Zr(1-x)/2Oy(x=0,0.1,0.2,0.4)。
1.2 催化劑性能評價
NO與H2的產氨反應在固定床流動反應器中進行,將經過壓片處理的催化劑置于石英管反應器中,反應器置于程序升溫管式爐內,爐溫由PID控制器進行調節。反應器中的進氣反應物由500×10-6NO,2 000×10-6H2及平衡氣N2組成,控制空速為30 000 h-1。待反應器出口處氣體成分穩定后,出口氣體中的NO,NO2及NH3濃度通過FTIR光譜儀(Thermo Nicolet 10)以1 Hz的采樣率進行測量。催化劑的性能評價通過NO向NH3的轉化率(SNH3)來表示[13],其定義如下:
SNH3= [NH3]out/[NO]in。
式中:[NH3]out為出口處的氨濃度;[NO]in為入口處一氧化氮濃度。
1.3 催化劑性能表征
使用Quantachrome NOVA 2000e自動氣體吸附儀來測量樣品的比表面積。使用N2作為吸附氣在77 K下進行吸附。在進行樣品比表面積測試之前,將樣品在300 ℃的真空下脫氣4 h以除去雜質。比表面積通過BET法進行測定。使用CuKα輻射(λ=1.540 6 ?)通過粉末X射線衍射(XRD,D8 ADVANCE DA VINCI)分析樣品的晶體結構。
在20°~80°的2θ范圍內獲得了XRD曲線,并通過Scherrer方程確定了催化劑的晶體尺寸。在Kratos AXIS Ultra DLD光譜儀上使用X射線光電子能譜法(XPS)分析催化劑表面的元素組成。C 1s光譜從278至298 eV收集,Cu 2p從923至958 eV收集,Ce 3d從330至350 eV收集,O 1s從765至815 eV收集。在Micromeritics Chemisorb 2720設備上進行了H2-TPR測試,以研究樣品的還原性。首先將樣品在600 ℃的N2中處理1 h,然后冷卻至室溫。H2-TPR在100~800 ℃的溫度下以10 ℃/min的斜率暴露于5%H2/N2的氣體混合物中,采用GC 2014氣相色譜儀和TCD檢測器監測氫氣含量變化。使用MAIA3 model 2016高分辨掃描電子顯微鏡來測試催化劑表面的光譜發射情況。采用Nicolet 6700型紅外光譜儀,通過原位漫反射紅外傅里葉變換技術(in-situ FTIR) 分別探究催化劑表面NO吸附情況及催化劑表面的反應過程。
2 結果與討論
2.1 催化劑性能
催化劑的產氨效率曲線見圖1。由圖1可見,當不摻雜銅時,沒有NO轉化為NH3。摻雜銅后,Cu負載對產氨效率的提高具有顯著作用,催化活性順序從大到小依次為x=0.1,x=0.2 ,x=0.4,隨著Cu摻雜量的增加,催化劑活性先增高后降低。當摻雜比例x=0.1時,在250~350 ℃溫度窗口內,催化劑能實現NO到NH3的全部轉化;對于摻雜比例x=0.2的催化劑,其活性相對降低,在275~350 ℃的溫度窗口內轉化率超過92%;摻雜比例x=0.4時,催化活性進一步降低,催化活性在250~350 ℃區間接近90%。結果表明,催化劑的產氨效率并不完全取決于摻雜量,而是取決于催化劑的結構、表面組成、氧空位及酸性位點等因素[14]。
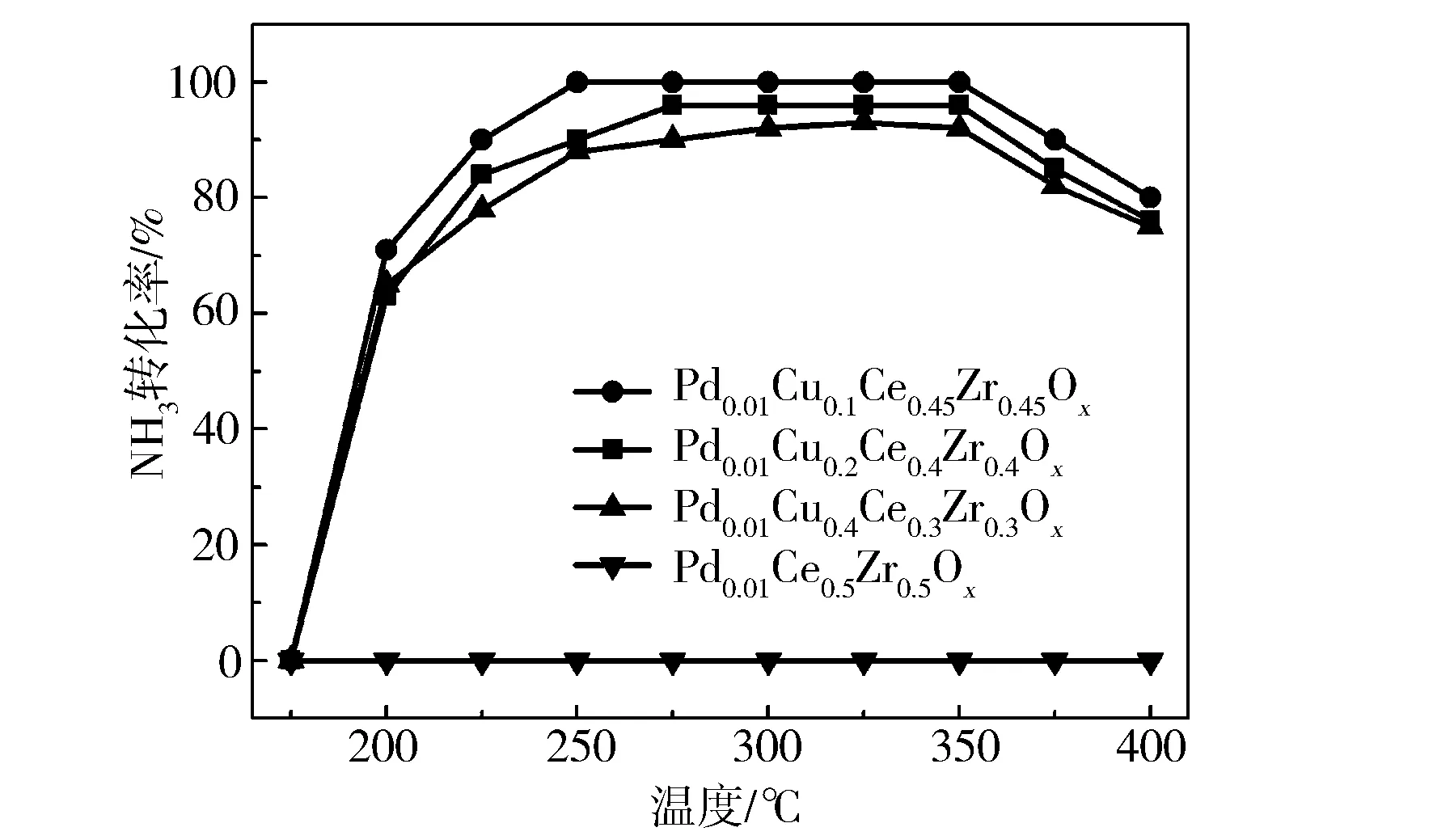
圖1 NO在不同催化劑上的氨轉化效率
2.2 催化劑表征
不同比例的Pd0.01CuxCe(1-x)/2Zr(1-x)/2Oy(x=0,0.1,0.2,0.4)催化劑的XRD圖譜見圖2。純Ce0.5Zr0.5O2樣品的反射主要位于29.25°,33.70°,34.03°,48.725°,48.90°和58.02°,分別對應于(1 0 1),(0 0 2),(1 1 0),(1 1 2),(2 0 0)和(2 1 1)Ce-Zr-O固溶體的平面。隨著銅含量的增加,衍射峰向更高的方向偏移。在Cu負載量x=0.1的樣品輪廓上未觀察到Cu或CuO峰。這表明銅氧化物的良好分散和固溶體的形成將增強銅氧化物與鈰鋯載體Ce0.5Zr0.5O2之間的相互作用,并促進反應的發生[15]。此外,由表1可見,催化劑的晶粒尺寸略有降低,這歸因于配位的Cu+和Cu2+的尺寸比Ce3+或Ce4+的尺寸小,以及銅氧化物與載體Ce0.5Zr0.5O2晶格中的結合。對于Cu負載量為x=0.2及x=0.4的樣品,檢測到35.4°和38.9°處發生了弱反射,這歸因于塊狀CuO的存在,這對催化活性沒有好處[16]。

圖2 不同催化劑的XRD譜圖
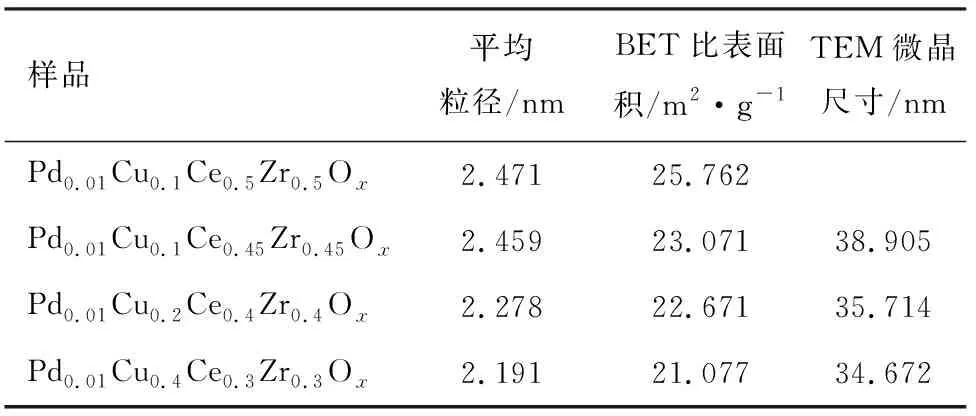
表1 不同催化劑的物理特征
圖3示出了不同比例的Pd0.01CuxCe(1-x)/2Zr(1-x)/2Oy(x=0,0.1,0.2,0.4)催化劑樣品的TEM圖像。從圖中可以看到很多顏色較深的黑點,這表明鈀物種的存在。隨著銅摻雜比例的增加,黑點逐漸聚集成大片的黑塊,表明鈀的聚集對催化活性不利[17]。當銅摻雜比例小于0.1時,鈀物種在催化劑表面高度分散。綜上所述,銅的摻雜會改變鈀的分散性,從而在一定程度上影響催化劑的催化活性。樣品的平均粒徑隨著銅的摻雜而略有降低,這表明將銅離子摻入螢石晶格有助于減小晶體尺寸[18]。這可以歸因于6個配位的Cu2+(0.073 nm)與Ce4+(0.10 nm)和Zr4+(0.084 nm)相比半徑較小。樣品的HRTEM形態見圖4。間隔值為0.310 nm和0.306 nm的晶格條紋分別對應于(1 1 1)平面的Ce0.5Zr0.5O2和Cu-Ce-Zr-O固溶體。在銅含量為x=0.1的樣品上未觀察到CuO的晶格條紋,這可能是由于Cu含量低所致。間隔為0.230 nm的晶格條紋對應于(1 1 1)平面的CuO(見圖4b和圖4c)。

圖4 不同催化劑的HRTEM圖像
結果表明催化劑中的Cu主要以Cu氧化物和Cu-Ce-Zr-O固溶體兩相存在[19]。Cu摻入Ce0.5Zr0.5O2受限制,高的Cu負載量將導致催化劑表面上形成大量的氧化銅,這在產氨反應中的活性較低,導致Cu負載量x=0.2及x=0.4的催化性能較差。
2.3 XPS光譜
圖5示出Pd0.01CuxCe(1-x)/2Zr(1-x)/2Oy(x=0,0.1,0.2,0.4)催化劑樣品的XPS分峰光譜。圖5a反映了Pd0.01CuxCe(1-x)/2Zr(1-x)/2Oy(x=0,0.1,0.2,0.4)樣品的Ce 3d光譜。光譜可分為8個不對稱峰,883.9 eV,888.9 eV,897.8 eV,903.1 eV,907.8 eV和916.7 eV對應的峰為Ce4+特征峰,885.6 eV和903.8 eV對應的峰為Ce3+特征峰。Ce3+與Ce4+的比例可反映催化劑表面氧空位情況,這與催化劑性能高度關聯[20]。由表2可見,在Cu負載量x=0.1和x=0.2時,表面Ce3+與Ce4+的比例與Cu負載量沒有直接關系。Cu負載量超過x=0.2時,Ce3+的含量降低,這可能是由于高分散性CuO的形成使催化劑表面的Ce3+再氧化,從而降低了Ce3+含量[21]。
圖5b 示出了Pd0.01CuxCe(1-x)/2Zr(1-x)/2Oy(x=0,0.1,0.2,0.4)催化劑樣品的Cu 2p光譜。可以將Cu XPS信號分解為以934.3 eV為中心的Cu 2p 3/2和以954.1 eV為中心的Cu 2p 1/2的兩個不對稱峰。在934.0 eV和953.7 eV處的主峰可以識別為Cu2+,而在940.1~945.1 eV范圍內的搖動峰歸因于Cu+。Cu+和Cu2+的同時存在可歸因于分散的CuO氧化催化劑表面上的Ce3+形成Cu+[22]。隨著Cu摻雜量的增加形成大量的CuO,使得Cu2+在Cu負載量x=0.4樣品中要低得多。
圖5c示出的O 1s光譜可以擬合到兩個主峰中。在529.6~529.9 eV范圍內的氧對應于來自催化劑晶格的氧(OL),而另一個以531.0~531.5 eV為中心的氧來自于表面化學吸附的氧(OA)。在催化劑上,OA與(OL+OA)的比率隨著Cu負載量的增加而減少。這表明在晶格中摻入Cu促進了銅離子與表面吸附氧OA的結合[23],從而使OA量減少。OA的減少降低了催化劑表面的氧化還原能力,降低了催化劑樣品對該反應的催化活性。

圖5 不同催化劑的XPS譜圖

表2 不同催化劑表面的原子組成
2.4 H2-TPR

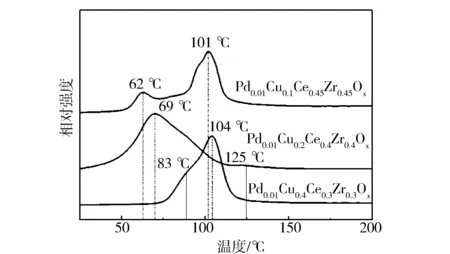
圖6 不同催化劑的H2-TPR譜圖
2.5 拉曼光譜
圖7示出Pd0.01CuxCe(1-x)/2Zr(1-x)/2Oy(x=0,0.1,0.2,0.4)催化劑樣品的拉曼光譜測試結果。在589,543,474,309,257,210 cm-1處的峰形特征為四方相結構Ce0.5Zr0.5O2的Raman振動峰[27]。隨著Cu負載量的增加,Ce0.5Zr0.5O2在257 cm-1和474 cm-1處的特征峰逐漸紅移,這表明引入Cu后形成Cu-Ce-Zr-O固溶體,使得Ce-Zr-O鍵長增加[28];此外,處于589 cm-1處的特征峰隨著Cu負載量的增加逐漸藍移,這可以看作是氧空位的產生[29]。這些結果表明,Cu2+可能結合到了Ce-Zr載體的晶格內。值得一提的是,對于Cu負載量為x=0.2及x=0.4的催化劑,在大約292 cm-1處出現了CuO的特征峰,而Cu負載量為x=0.1的催化劑特征峰并不明顯,這表明過量的CuO累積在催化物的表面,并與Cu-Ce-Zr-O固溶體產生了很強的相互作用。而這種過強的相互作用對產氨反應并沒有積極影響。

圖7 不同催化劑的拉曼譜圖
此外,632 cm-1和463 cm-1處的兩個峰面積(表示為A632和A463)的比值可反映出相對的表面氧空位數量[30]。不同催化劑的632 cm-1和463 cm-1的峰面積比擬合結果見表3。當Cu摻雜量為0.1時,A632/A463的值最高,表明催化劑表面存在較多的氧空位。催化劑的氧空位數量從大到小依次為Pd0.01Cu0.1Ce0.45Zr0.45Ox,Pd0.01Cu0.2Ce0.4Zr0.4Ox,Pd0.01Cu0.4Ce0.3Zr0.3Ox, Pd0.01Cu0.1Ce0.5Zr0.5Ox,與催化劑的活性變化一致。由此可見,Cu摻雜過多或過少都會對表面氧空位產生不利影響。眾所周知,氧空位的產生將促進銅和Ce0.5Zr0.5O2載體之間形成強大的金屬-載體相互作用。因此,當Cu的摻雜量為0.1時,氧空位的最大數量導致Cu與Ce0.5Zr0.5O2的相互作用更強,從而促進催化劑的轉化效率。

表3 不同催化劑的A632/A463比值
2.6 原位表征

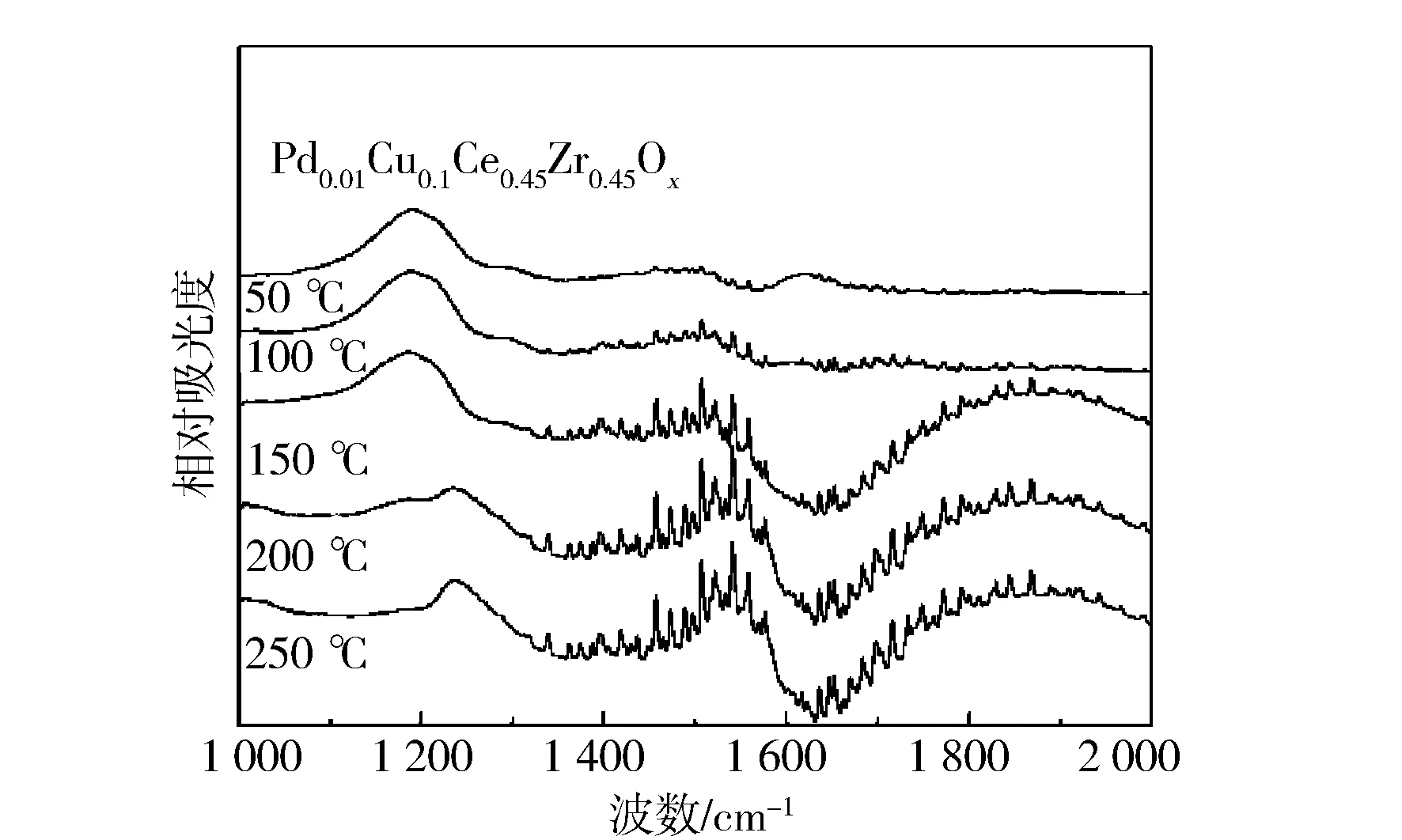
圖8 不同催化劑的NO吸附in-situ FTIR譜圖
此外,對Pd0.01Cu0.1Ce0.45Zr0.45Ox催化劑樣品的NO與H2反應性也進行了研究(見圖9)。
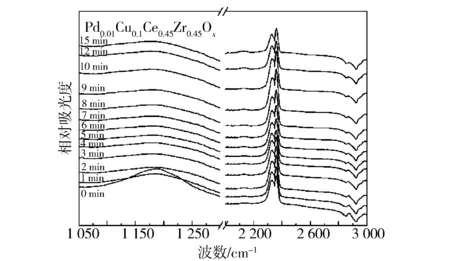
圖9 不同催化劑產氨反應的in-situ FTIR譜圖
在250 ℃下用NO預吸附飽和后,向催化劑通入H2,并測定其0~15 min的原位紅外光譜。當開始通入H2后,催化劑表面的各種硝酸鹽物種隨著H2通入時間的增加逐漸減少,在1 min時只剩下強度較弱的吸附峰,說明表面吸附的NOx物種以較快的反應速度消耗;在15 min時只剩下很弱的吸附峰,說明大部分吸附的NOx物種均可參與NO與H2的產氨反應[33]。
3 結論
a) 隨著Cu負載量的增多,催化劑表面的Ce3+含量減少,Ce4+的相對含量增加,而Cu2+相對含量減少,催化劑表面化學吸附氧的比例減少;結果表明,OA的含量及Cu2+與Ce4+的競爭關系是決定該反應的重要因素;
b) 銅與鈰鋯之間的過強相互作用,使得隨著Cu負載量的增加,CuO在催化劑中形成,對反應不利,過多的Cu負載量抑制了Pd0.01CuxCe(1-x)/2Zr(1-x)/2Oy催化劑的低溫還原能力;
c) 拉曼圖譜部分特征峰的藍移及紅移,在催化劑表面產生了更多的氧空位;過量的CuO累積在催化物的表面,并與Cu-Ce-Zr-O固溶體產生了很強的相互作用,對反應不利;
d) 酸度在該反應中也起到重要的作用;銅摻雜可以產生更多的酸性位點,有效地改善了NO的活化,從而促進NO向NH3的轉化;Pd0.01Cu0.1Ce0.45Zr0.45Ox催化劑樣品的氧化還原能力強,但其對NOx的吸附能力較弱,且該反應的反應速率較高;
e) Cu的摻雜改善了催化劑表面物種的分布,提高了活性物質的分散度,增強了催化劑的催化還原能力,并增加表面化學吸附氧及催化劑表面的酸性位點,從而在多方面改善了催化劑的性能,這種新式催化劑有希望成為改進TWC催化劑實現被動SCR脫附NOx的一個非常有效的方案。
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
