玫瑰紅景天活性成分絡塞維的合成研究
2021-07-06 14:05:26鐘婷胡亞丹蘇進楊淑珍何貝橋張園園
中國現代中藥 2021年5期
鐘婷,胡亞丹,蘇進,楊淑珍,何貝橋,張園園
北京中醫藥大學 中藥學院,北京 102488
絡塞維為肉桂醇基-6-O-(α-L-吡喃阿拉伯糖基)-β-D-吡喃葡萄糖苷(圖1),是由肉桂醇與蠶豆糖所構成的二糖苷。絡塞維是天然抗氧化中藥玫瑰紅景天RhodiolaroseaL.的活性成分和指標性成分[1]。據文獻報道,其具有抗疲勞、抗輻射、抗氧化、抗癌、增強免疫、改善學習記憶能力等多種藥理活性[2-5],應用前景廣闊。絡塞維主要是從玫瑰紅景天中提取、分離、純化后制備[6],但質量分數僅為0.21%~0.55%[1],使得分離純化難度大、成本高,有必要進行化學合成。目前,絡塞維的合成方法主要有2種[7-9]:1)Msashi等[7]將D-葡萄糖與丙烯醇通過固定化β-葡萄糖苷酶催化成苷,然后通過保護與脫保護策略,得到烯丙基-2,3,4-三-O-苯甲酰基-β-D-吡喃葡萄糖苷,再與溴代阿拉伯糖反應,最后與苯基硼酸反應,脫苯甲酰基保護基得絡塞維。但該方法使用的丙烯醇為劇毒、管制藥品,醋酸鈀和固定化β-葡萄糖苷酶價格昂貴,且需經過2次苯甲酰基保護的二糖苷的分離,操作煩瑣。2)另一種思路是通過糖6位羥基暴露的D-吡喃葡萄糖受體與L-吡喃阿拉伯糖供體合成二糖中間體,再將其制備成二糖供體與肉桂醇進行糖苷化反應,最后脫保護基得絡塞維。但是在二糖中間體合成過程中副產物多,分離困難,且進一步制備二糖供體時收率大大降低,很難得到。
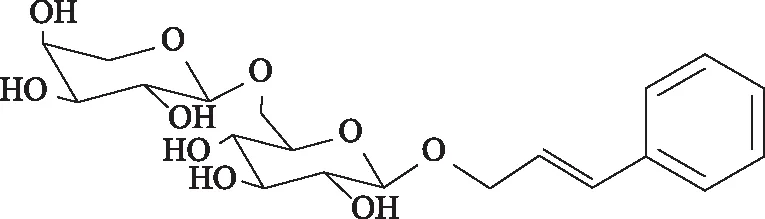
圖1 絡塞維結構
本研究以肉桂醇為原料,與全乙酰-α-D-溴代葡萄糖進行成苷反應,經脫乙酰基保護獲得肉桂醇基-β-D-吡喃葡萄糖苷,接著用三苯基氯甲烷對糖的6′位羥基選擇性保護、其他位羥基乙酰化保護及脫糖6′位保護基,特異性地將糖6′位伯羥基暴露,再與全乙酰-β-L-溴代吡喃阿拉伯糖成苷,最后脫乙酰保護基得到絡塞維,路線見圖2。本方法充分考慮糖類化合物合成過程中的區域選擇性和立體選擇性,收率較高,且未見文獻報道。獲得的產物和關鍵中間體可用于相關化合物的深入研究。本研究還可為苯丙素糖苷類化合物的制備提供參考,有助于該類天然產物在醫藥領域的進一步應用。

注:1.1,2,3,4,6-五-O-乙酰基-α-D-吡喃葡萄糖;2.2,3,4,6-四-O-乙酰基-α-D-溴代吡喃葡萄糖;3.肉桂醇基-2,3,4,6-四-O-乙酰基-β-D-吡喃葡萄糖苷;4.肉桂醇基-O-β-D-吡喃葡萄糖苷;5.肉桂醇基-6-O-三苯基甲基-β-D-吡喃葡萄糖苷;6.肉桂醇基-2,3,4-三-O-乙酰基-6-O-三苯基甲基-β-D-吡喃葡萄糖苷;7.肉桂醇基-2,3,4-三-O-乙酰基-β-D-吡喃葡萄糖苷;8.肉桂醇基-2,3,4-三-O-乙酰基-6-O-(2,3,4-三-O-乙酰基-α-L-吡喃阿拉伯糖基)-β-D-吡喃葡萄糖苷;9.絡塞維。圖2 絡塞維的合成路線
1 材料
ASCEND-400型核磁共振儀(瑞士Bruker公司);WZZ-2B型半自動旋光儀(上海精密科學儀器有限公司);RE-52AA型旋轉蒸發儀(上海亞榮生化儀器廠);JM-B5002型電子天平(余姚市紀銘稱重校驗設備有限公司);X-5型顯微熔點測定儀(鞏義市科瑞儀器有限公司);LC-20AT型制備液相色譜儀(日本島津公司);Cosmosil 5C18AR-Ⅱ型制備色譜柱(250 mm×10 mm,5 μm,日本Nacalai Tesque公司)。
濃硫酸、乙酸酐(純度:97%)、分析純甲醇(北京化工廠);甲醇鈉(純度:98%,Acros Organics公司);33%溴化氫乙酸溶液(33% HBr-CH3COOH)、三苯基氯甲烷(純度:98%,北京伊諾凱科技有限公司);Ag2CO3(純度:99%,天津福晨化學試劑廠);肉桂醇(純度:98%,阿拉丁試劑上海有限公司);D-葡萄糖、L-阿拉伯糖(純度均為98%,阿達瑪斯試劑有限公司);薄層色譜(TLC)板G(青島海洋化工有限公司)。
2 方法與結果
2.1 1,2,3,4,6-五-O-乙酰基-α-D-吡喃葡萄糖(1)的合成
取乙酸酐11.5 mL、濃硫酸30 μL,置于圓底燒瓶中,冰浴條件下,加入D-葡萄糖4.0 g,攪拌反應1 h,撤除冰浴,室溫繼續反應3 h,TLC監測至反應完全。反應液用二氯甲烷50 mL萃取3次,收集、合并有機相并用水洗至中性,無水硫酸鈉干燥,濾除硫酸鈉后,將有機相減壓濃縮得白色粉末,收率為93.7%,熔點為112~114 ℃,與文獻報道一致[10]。
2.2 2,3,4,6-四-O-乙酰基-α-D-溴代吡喃葡萄糖(2)的合成
取化合物18.0 g,用二氯甲烷10 mL攪拌溶解,加入33%HBr-CH3COOH溶液5 mL,室溫反應2~3 h,TLC監測至反應完全。反應液分散于適量冰水中,收集有機相,水相用二氯甲烷15 mL萃取3次,合并有機相并用水洗至中性,無水硫酸鈉干燥,濾除硫酸鈉后,減壓回收溶劑,甲基叔丁基醚結晶,得白色晶體,收率為87.6%,熔點為86~88 ℃,與文獻報道一致[11]。
2.3 肉桂醇基-2,3,4,6-四-O-乙酰基-β-D-吡喃葡萄糖苷(3)的合成
取肉桂醇1.3 g,用二氯甲烷20 mL攪拌溶解,加入Ag2CO32.0 g、化合物26.6 g,避光室溫反應過夜,TLC監測至反應完全,抽濾,濾餅用二氯甲烷洗1次,合并濾液,減壓回收溶劑,得粗產物,直接投入2.4項下反應。
2.4 肉桂醇基-O-β-D-吡喃葡萄糖苷(4)的合成
將化合物3粗產物以5倍量甲醇于圓底燒瓶中攪拌溶解,加入適量甲醇鈉調pH 8~9,室溫反應4 h,TLC監測至反應完全,加適量陽離子交換樹脂調pH 6~7,濾除陽離子交換樹脂,減壓回收溶劑,柱色譜分離(二氯甲烷-甲醇,12∶1),得白色粉末,收率為54.7%,熔點為98~99 ℃。1H-NMR(CD3OD,400 MHz)δ:7.40 (2H,d,J=7.2 Hz,H-5,9),7.28(2H,t,J=7.6 Hz,H-6,8),7.20(1H,t,J=7.2 Hz,H-7),6.67(1H,d,J=16.0 Hz,H-3),6.37(1H,dt,J=6.0,16.0 Hz,H-2),4.55(1H,dd,J=13.2,5.6 Hz,H-1),4.43(1H,d,J=7.6 Hz,H-1′),4.33(1H,dd,J=12.4,6.4 Hz,H-1),3.93(1H,dd,J=11.6,1.6 Hz,H-6′),3.78(1H,dd,J=12.0,5.2 Hz,H-6′),3.52~3.30(4H,m,H-2′~5′)。13C-NMR(CD3OD,100 MHz)δ:136.8(C-4),132.5(C-3),128.4(C-6,8),127.5(C-7),126.3(C-5,9),125.4(C-2),102.0(C-1′),76.7(C-3′),76.5(C-5′),73.7(C-2′),70.2(C-4′),69.6(C-1),61.5(C-6′)。以上數據與文獻報道一致[12]。
2.5 肉桂醇基-6-O-三苯基甲基-β-D-吡喃葡萄糖苷(5)的合成
參考文獻[13]方法,取化合物41.0 g置于圓底燒瓶中,用吡啶5 mL攪拌溶解,加入三苯基氯甲烷1.4 g,于50 ℃下反應7 h,TLC監測至反應完全,降溫至室溫后投入2.6項下反應。
2.6 肉桂醇基-2,3,4-三-O-乙酰基-6-O-三苯基甲基-β-D-吡喃葡萄糖苷(6)的合成
向2.5項下體系中加入乙酸酐1.9 mL、4-二甲氨基吡啶(DMAP)20 mg,室溫攪拌過夜,TLC監測至反應完全,將反應液分散于適量冰水中,用二氯甲烷10 mL萃取3次,合并有機相,冰水和適量的稀鹽酸各洗2次至弱酸性,最后用冰水洗至中性,無水硫酸鈉干燥,濾除硫酸鈉后,減壓回收溶劑,柱色譜分離(石油醚-乙酸乙酯,5∶1),得黃色糖漿狀物質,收率為76.4%。1H-NMR (DMSO-d6,400 MHz)δ:7.44 (8H,d,J=7.6 Hz,H-5,9,3″,7″,9″,13″,15″,19″),7.34(8H,dd,J=7.2,14.8 Hz,H-6,8,4″,6″,10″,12″,16″,18″),7.26 (4H,t,J=7.2 Hz,H-7,5″,11″,17″),6.68(1H,d,J=16 Hz,H-3),6.41(1H,dt,J=5.6,16.0 Hz,H-2),5.25(1H,t,J=9.6 Hz,H-3′),5.18(1H,t,J=9.6 Hz,H-4′),4.96(1H,t,J=8.0 Hz,H-2′),4.92(1H,d,J=8.0 Hz,H-1′),4.51(1H,dd,J=5.2,13.2 Hz,H-1),4.36(1H,dd,J=6.0,13.6 Hz,H-1),3.91~2.87(3H,m,H-5′,6′),2.05(3H,s,glc-4′-OAc),1.95(3H,s,glc-3′-OAc),1.72(3H,s,glc-2′-OAc)。13C-NMR(DMSO-d6,100 MHz)δ:170.1(glc-2′-OAc),169.6(glc-3′-OAc),169.1(glc-4′-OAc),143.9(C-2″,8″,14″),136.7(C-4),132.4(C-3),129.1(C-6,8),128.7(C-4″,6″,10″,12″,16″,18″),128.4~128.3(C-7,3″,7″,9″,13″,15″,19″),127.5(C-5″,11″,17″),126.9(C-5,9),126.0(C-2),99.3(C-1′),86.3(Ph3-C),73.0(C-3′),72.4(C-5′),71.7(C-2′),69.4(C-4′),68.6(C-1),61.8(C-6′),20.9(glc-4′-OAc),20.8(glc-2′-OAc),20.6(glc-3′-OAc)。
2.7 肉桂醇基-2,3,4-三-O-乙酰基-β-D-吡喃葡萄糖苷(7)的合成
取化合物61.0 g用二氯甲烷10 mL攪拌溶解,加入冰醋酸2.5 mL,于-20 ℃下滴加33% HBr-CH3COOH溶液195.8 μL,并在此溫度下繼續反應2 h,TLC監測至反應完全。將反應液分散于適量冰水中,收集有機相,水相用二氯甲烷5 mL萃取3次,合并有機相,并用冰水洗至中性,無水硫酸鈉干燥,濾除硫酸鈉后,減壓回收溶劑,柱色譜分離(石油醚-乙酸乙酯,2∶1),得無色糖漿狀物質,收率為70.8%。1H-NMR (CDCl3,400 MHz)δ:7.38(2H,d,J=7.2 Hz,H-5,9),7.33(2H,t,J=7.2 Hz,H-6,8),7.26(1H,t,J=7.2 Hz,H-7),6.60(1H,d,J=16.0 Hz,H-3),6.23(1H,dt,J=6.4,16.0 Hz,H-2),5.27(1H,t,J=9.6 Hz,H-3′),5.07(1H,t,J=9.6 Hz,H-4′),4.99(1H,dd,J=8.0,9.6 Hz,H-2′),4.66(1H,d,J=8.0 Hz,H-1′),4.54~3.53(5H,m,H-1,5′,6′),2.12(3H,s,glc-2′-OAc),2.07(3H,s,glc-4′-OAc),2.06(3H,s,glc-3′-OAc)。13C-NMR(CDCl3,100 MHz)δ:171.7(glc-2′-OAc),170.3(glc-3′-OAc),169.6(glc-4′-OAc),136.4(C-4),133.0(C-3),128.7(C-6,8),128.0(C-7),126.5(C-5,9),124.7(C-2),99.7(C-1′),75.5(C-3′),74.1(C-5′),71.5(C-2′),69.9(C-4′),68.9(C-1),63.0(C-6′),20.8(glc-2′-OAc),20.8(glc-4′-OAc),20.6(glc-3′-OAc)。
2.8 肉桂醇基-2,3,4-三-O-乙酰基-6-O-(2,3,4-三-O-乙酰基-α-L-吡喃阿拉伯糖基)-β-D-吡喃葡萄糖苷(8)的合成
取化合物70.4 g,用二氯甲烷6 mL攪拌溶解,加入Ag2CO30.2 g、2,3,4-三-O-乙酰基-β-L-溴代吡喃阿拉伯糖0.5 g(制備同2.1、2.2項下方法),避光室溫反應過夜,TLC監測至反應完全,抽濾,濾餅用二氯甲烷洗1次,減壓蒸干溶劑,制備液相色譜法分離(甲醇-水,7∶3),得化合物8,收率為62.7%。1H-NMR(CDCl3,400 MHz)δ:7.32(2H,d,J=7.2 Hz,H-5,9),7.26(2H,t,J=7.2 Hz,H-6,8),7.18 (1H,t,J=7.2 Hz,H-7),6.55(1H,d,J=16.0 Hz,H-3),6.37(1H,dt,J=5.2,15.6 Hz,H-2),5.18~4.85(6H,m,H-2′~4′,2″~4″),4.52(1H,d,J=8.0 Hz,H-1′),4.41(1H,d,J=6.8 Hz,H-1″),4.45~4.40(1H,m,H-1),4.21(1H,dd,J=6.4,13.2 Hz,H-1),3.96~3.49(5H,m,H-5′,5″,6′),2.06(3H,s,ara-4″-OAc),1.99(3H,s,glc-4′-OAc),1.98(3H,s,ara-3″-OAc),1.97(3H,s,glc-3′-OAc),1.95(3H,s,glc-2′-OAc),1.92(3H,s,ara-2″-OAc)。13C-NMR(CDCl3,100 MHz)δ:170.2(glc-2′-OAc),170.2(glc-3′-OAc),170.1(ara-2″-OAc),169.5(ara-3″-OAc),169.4(glc-4′-OAc),169.4(ara-4″-OAc),136.4(C-4),133.0(C-3),128.6(C-6,8),127.9(C-7),126.5(C-5,9),124.4(C-2),100.8(C-1″),99.3(C-1′),73.2(C-3′),72.9(C-5′),71.4(C-2′),70.0(C-3″),69.5(C-2″),69.1(C-4′),69.0(C-1),67.9(C-4″),67.5(C-6′),63.1(C-5″),20.9(glc-2′-OAc),20.8(ara-2″-OAc),20.7(glc-4′-OAc),20.7(ara-4″-OAc),20.6(glc-3′-OAc),20.6(ara-3″-OAc)。
2.9 絡塞維(9)的合成

3 討論
3.1 化合物1的合成
以葡萄糖為原料、濃硫酸為催化劑、乙酸酐為酰化試劑,合成全乙酰-α-D-吡喃葡萄糖。在D-葡萄糖投料4.0 g、濃硫酸用量為30 μL的條件下,以室溫反應完全所需時間和收率為指標,考察葡萄糖-乙酸酐投料比分別為1∶3.5、1∶4.5、1∶5.5、1∶7.0。結果表明,化合物1收率分別為65.6%、91.9%、93.7%、94.6%,反應時間分別為18.0、5.0、3.0、2.5 h。綜合成本和效率,以葡萄糖-乙酸酐1∶5.5為最佳條件。
3.2 化合物2的合成
用33% HBr-CH3COOH溶液對化合物1進行溴代反應,葡萄糖1.5 g用33% HBr-CH3COOH溶液1 mL反應完全,且產率較高。溴代糖常溫易分解,后處理需在低溫下進行,用甲基叔丁基醚結晶。
3.3 化合物4的合成
將2.3項下粗產物在堿性條件下與甲醇進行酯交換反應,不同用量的甲醇對反應完成時間和收率有影響,甲醇用量增加,反應時間縮短,產率提高。綜合成本和效率,選用5倍量甲醇進行反應。
3.4 化合物8的合成
化合物7在Ag2CO3催化下與2,3,4-三-O-乙酰基-β-L-溴代吡喃阿拉伯糖進行親核取代反應,合成糖苷,考察各反應物的投料比對產物收率的影響。結果表明,隨著Ag2CO3和溴代阿拉伯糖投料量的增加,反應收率增加,但變化幅度不大。從節約成本的角度,選擇化合物7-Ag2CO3-三乙酰溴代阿拉伯糖投料比為1∶0.75∶1.5,收率為62.7%。
3.5 化合物9的合成
化合物9的合成同化合物4的合成,用甲醇和甲醇鈉脫乙酰基保護,但為了防止脫保護過程中二糖鏈斷開,應加大甲醇的用量(10倍量),使其在短時間內快速反應完全。
4 結論
近年來,研究者們逐漸認識到寡糖及糖苷類化合物的生物學意義,寡糖類化合物的化學合成技術也快速發展。大量的天然產物的糖基部分存在龍膽二糖、蕓香糖、蠶豆糖等1→6連接二糖,但是合成報道較少,因此,探索區域選擇性和立體選擇性高的路線合成1→6連接二糖具有一定的研究意義。
本研究在合成絡塞維的過程中,用三苯基氯甲烷對葡萄糖的6′位伯羥基進行選擇性保護,脫保護后6′位羥基裸露從而合成1→6連接糖苷鍵。三苯基氯甲烷基團龐大,使用其作保護基團具有選擇性高、保護基團產物穩定和保護基團容易脫去的優點。本研究使用三苯基氯甲烷作為保護基團合成絡塞維具有創新性,以期能夠為相關二糖的合成研究工作提供新思路。
絡塞維有廣泛的生物學活性,本研究制備糖基供體后,在成苷反應中通過7步合成絡塞維,總產率為15.92%。與文獻相比,雖然未能縮短合成步驟,但實驗成本降低,且條件溫和、操作簡單,對人體的危害和環境的污染影響較小,具有良好的工業化前景。需要說明的是,在化合物8的合成過程中,由于樣品量較少,采用了制備液相進行分離,如后期大量制備可進一步探索硅膠柱色譜法進行分離。
