高效液相色譜-串聯(lián)質(zhì)譜法測定動物源性食品中噻節(jié)因殘留
2021-07-21 13:21:04肖泳周叢鄧航馮燕英潘照唐吉旺袁列江
食品與發(fā)酵工業(yè) 2021年13期
肖泳,周叢,鄧航,馮燕英,潘照,唐吉旺,袁列江
(湖南省產(chǎn)商品質(zhì)量監(jiān)督檢驗(yàn)研究院,湖南 長沙,410007)
噻節(jié)因(dimethipin)是一種植物生長調(diào)節(jié)劑,可促進(jìn)植物成熟,使玉米、苗木、橡膠樹和葡萄落葉,并能降低收獲后水稻和向日葵種子的含水量。噻節(jié)因具有中等毒性,長期使用可能對生態(tài)和食品安全產(chǎn)生較大風(fēng)險(xiǎn)。美國環(huán)保局將噻節(jié)因列為可能的人類致癌物,目前歐盟禁止噻節(jié)因在農(nóng)作物上使用,國際食品法典委員會、美國、日本等國家或組織制定了食品中噻節(jié)因的最大殘留限量。我國食品安全國家標(biāo)準(zhǔn)(GB 2763—2019)中也制定了油料和油脂、馬鈴薯、哺乳動物肉類及內(nèi)臟(海洋哺乳動物除外)、禽肉類、禽類內(nèi)臟、蛋類、生乳中噻節(jié)因的最大殘留限量值,但該標(biāo)準(zhǔn)中哺乳動物肉類及內(nèi)臟(海洋哺乳動物除外)、禽肉類、禽類內(nèi)臟、蛋類、生乳中噻節(jié)因的測定是參照蜂蜜(GB/T 20771—2008)的方法。
目前,國內(nèi)外關(guān)于食品中噻節(jié)因的檢測方法主要有氣相色譜法[1]、液相色譜法[1-2]、氣質(zhì)聯(lián)用法[1,3-11]、液質(zhì)聯(lián)用法[12-14]。氣相色譜法和氣質(zhì)聯(lián)用法前處理較繁瑣、耗時(shí)較長;液相色譜法靈敏度低;液質(zhì)聯(lián)用法前處理相對簡便且具有高特異性和高靈敏度,但現(xiàn)有文獻(xiàn)方法大多針對果蔬類樣品,對動物源性食品中噻節(jié)因檢測的研究較少。本文以動物源性食品為研究對象,采用內(nèi)標(biāo)法定量,建立了高效液相色譜-串聯(lián)質(zhì)譜法測定動物源性食品中噻節(jié)因殘留的分析方法,并研究了離子源、色譜柱、流動相、提取溶劑、凈化方式、固相萃取柱等因素對噻節(jié)因的離子化程度、提取效率及凈化效果的影響。
1 實(shí)驗(yàn)部分
1.1 儀器、試劑與材料
Vanquish-TSQ Quantis高效液相色譜-串聯(lián)質(zhì)譜儀,美國Thermo公司;MV5多通道氮吹濃縮儀、DHS-220雙模式自動均質(zhì)儀,北京萊伯泰科有限公司;Allegra 64R高速冷凍離心機(jī),美國Beckman Coulter公司;GM200刀式研磨儀,德國Retsch公司;Venusil MP C18(2)色譜柱(150 mm×2.1 mm,3 μm),天津博納艾杰爾科技有限公司;C18固相萃取柱(2 g,12 mL),北京中檢維康技術(shù)有限公司。
噻節(jié)因(純度99.0%)、4-羥基苯甲酸異丁酯(純度99.0%),德國Dr.Ehrenstorfer公司;乙腈、正己烷、甲酸、乙酸,均為HPLC級,上海安譜公司;MgSO4、NaCl,均為分析純,國藥集團(tuán)有限公司。
稱取適量噻節(jié)因和4-羥基苯甲酸異丁酯標(biāo)準(zhǔn)品,分別用乙腈配制成1.0 mg/mL的標(biāo)準(zhǔn)儲備液(噻節(jié)因可加少量丙酮溶解再用乙腈定容),于0~4 ℃冰箱中儲存。根據(jù)需要將噻節(jié)因標(biāo)準(zhǔn)儲備液用乙腈稀釋成適當(dāng)濃度的標(biāo)準(zhǔn)工作液,將4-羥基苯甲酸異丁酯內(nèi)標(biāo)儲備液用乙腈稀釋成100 ng/mL的內(nèi)標(biāo)工作液,于0~4 ℃冷藏儲存,備用。
1.2 實(shí)驗(yàn)方法
1.2.1 樣品制備
豬、牛、羊、雞等畜禽類樣品取可食肌肉組織、內(nèi)臟約200 g,切碎后經(jīng)組織搗碎機(jī)搗碎均勻,裝入潔凈容器內(nèi),密封,于-20 ℃以下冷凍保存;蛋類取10枚,去殼后經(jīng)組織搗碎機(jī)充分混勻,裝入潔凈容器內(nèi),密封,于-20 ℃以下冷凍保存;生乳樣品取約200 g充分混勻,裝入潔凈容器內(nèi),密封,于-20 ℃以下冷凍保存。
1.2.2 樣品前處理
稱取5 g(精確至0.01 g)制備均勻的樣品,置于50 mL具塞離心管中,加入5 mL水(生乳不用加水),準(zhǔn)確加入50 μL內(nèi)標(biāo)工作溶液(100 ng/mL),混勻靜置20 min;加入25 mL體積分?jǐn)?shù)1%的乙酸乙腈溶液,15 000 r/min均質(zhì)2 min,加入1~2 g NaCl、2~3 g MgSO4,快速混勻2 min,7 000 r/min離心5 min,取15 mL提取液,用2×10 mL乙腈飽和的正己烷脫脂2次,準(zhǔn)確移取其中10 mL提取液于40 ℃氮吹濃縮至約1 mL,待進(jìn)一步凈化。
用10 mL乙腈活化C18固相萃取柱,上樣,用10 mL乙腈洗脫,接收提取液和洗脫液,于40 ℃氮吹濃縮至近干,用1.0 mL乙腈-水溶液[V(乙腈)∶V(水)=3∶2]溶解殘?jiān)?過0.22 μm有機(jī)濾膜后供高效液相色譜-串聯(lián)質(zhì)譜儀測定。
1.2.3 色譜和質(zhì)譜條件
色譜柱Venusil MP C18(2)柱(150 mm×2.1 mm,3 μm);柱溫40 ℃;流速0.4 mL/min;流動相A為水,流動相B為乙腈。梯度洗脫程序:0~1.00 min,10%B~60%B;1.00~3.50 min,60%B~95%B;3.50~4.00 min,95%B;4.00~4.01 min,95%B~10%B;4.01~5.00 min,10%B。進(jìn)樣體積10 μL。
離子源:大氣壓化學(xué)電離源(atmospheric pressure chemical ionization source,APCI);掃描模式:負(fù)離子掃描;掃描方式:多反應(yīng)監(jiān)測;離子傳輸管溫度310 ℃;蒸發(fā)溫度325 ℃;鞘氣流量8.4 L/min;輔助氣流量2.42 L/min;放電電流:5 μA;其他質(zhì)譜條件見表1。

表1 噻節(jié)因和4-羥基苯甲酸異丁酯的質(zhì)譜參數(shù)
2 結(jié)果與討論
2.1 質(zhì)譜條件的選擇
液質(zhì)聯(lián)用法檢測噻節(jié)因常用的離子源有電噴霧離子源(electron spray ionization, ESI)[13-14]和APCI源[12]。本方法配制10 mg/L的噻節(jié)因標(biāo)準(zhǔn)溶液和1 mg/L的4-羥基苯甲酸異丁酯內(nèi)標(biāo)溶液,注入ESI源和APCI源,并分別在正離子和負(fù)離子模式下進(jìn)行一級質(zhì)譜圖全掃描,確定母離子,然后對準(zhǔn)母離子進(jìn)行子離子掃描,不同模式下二級離子碎片質(zhì)譜圖見圖1。結(jié)果表明,噻節(jié)因在APCI源中的離子化效果要優(yōu)于ESI源,且負(fù)離子模式下的響應(yīng)明顯高于正離子模式,與楊濤等[12]的研究結(jié)果一致。因此,選擇APCI源負(fù)離子模式對噻節(jié)因和4-羥基苯甲酸異丁酯進(jìn)行離子化,并在多反應(yīng)監(jiān)測模式下優(yōu)化各種質(zhì)譜參數(shù),得到最佳質(zhì)譜條件,噻節(jié)因和4-羥基苯甲酸異丁酯的質(zhì)譜參數(shù)見表1。

a-噻節(jié)因:APCI-;b-噻節(jié)因:APCI+;c-噻節(jié)因:ESI-;d-4-羥基苯甲酸異丁酯:APCI-
2.2 色譜條件的選擇
色譜柱的選擇對樣品的分離十分重要,通常不同類型的填料對同一樣品有不同的保留效果。本文重點(diǎn)比較了Venusil MP C18(2)柱(150 mm×2.1 mm,3 μm)和Venusil HILIC柱(150 mm×2.1 mm,3 μm)對目標(biāo)化合物的保留情況,并采用同濃度的標(biāo)準(zhǔn)溶液考察了分別以乙腈-水、乙腈-0.1%甲酸水溶液和乙腈-10 mmol/L乙酸銨水溶液作流動相時(shí),對目標(biāo)化合物離子化程度的影響(圖2)。結(jié)果表明,采用MP C18(2)柱,以乙腈-水作為流動相時(shí),噻節(jié)因的響應(yīng)明顯高于其他2種流動相;采用HILIC柱,以乙腈-10 mmol/L乙酸銨水溶液作為流動相時(shí),噻節(jié)因的響應(yīng)明顯高于其他2種流動相。這2種色譜柱最佳流動相條件下噻節(jié)因的響應(yīng)相差不大,但是MP C18(2)柱對噻節(jié)因的保留效果要優(yōu)于HILIC柱,且流動相中不需要添加銨鹽。因此,選擇Venusil MP C18(2)柱作為噻節(jié)因的分離色譜柱,并以乙腈-水作為流動相進(jìn)行梯度洗脫。

a-色譜柱:MP C18(2),流動相:乙腈-水;b-色譜柱:MP C18(2),流動相:乙腈-0.1%甲酸水溶液;c-色譜柱:MP C18(2),流動相:乙腈-10 mmol乙酸銨水溶液;d-色譜柱:HILIC,流動相:乙腈-水;e-色譜柱:HILIC,流動相:乙腈-0.1%甲酸水溶液;f-色譜柱:HILIC,流動相:乙腈-10 mmol乙酸銨水溶液
2.3 提取和凈化條件的優(yōu)化
噻節(jié)因常用的提取溶劑有乙腈[13-14]、體積分?jǐn)?shù)1%的乙酸乙腈溶液[3-4,7]、丙酮[12]等。李南等[7]比較了正己烷、丙酮、純乙腈和1%的乙酸乙腈溶液對堅(jiān)果中農(nóng)藥的提取效果,最終得出1%的乙酸乙腈溶液效果最佳;張煜卓等[10]比較了乙腈、丙酮、乙酸乙酯、二氯甲烷對南果梨中農(nóng)藥的提取效果,最終得出乙腈效果最佳;楊濤等[12]比較了乙腈、甲醇、丙酮、水和丙酮-水等體積混合對果蔬中噻節(jié)因的提取效果,最終得出丙酮效果最佳。對于動物源性食品,由于脂肪含量高,丙酮提取液濃縮后呈黏稠狀液體,不利于后期凈化;乙腈和1%的乙酸乙腈溶液作為提取溶劑,方法回收率均可達(dá)80%以上,當(dāng)用正己烷對提取液進(jìn)行脫脂處理后,發(fā)現(xiàn)提取液明顯變得澄清,且不會影響噻節(jié)因的回收率;采用含1%的乙酸乙腈溶液作為提取溶劑,可以進(jìn)一步沉淀動物源性樣品中的蛋白質(zhì),使上機(jī)待測液較純乙腈作為提取溶劑時(shí)更澄清。因此,實(shí)驗(yàn)選擇體積分?jǐn)?shù)1%的乙酸乙腈溶液作為提取溶劑,并用正己烷對提取液進(jìn)行脫脂處理。
噻節(jié)因凈化常用的固相萃取柱有NH2柱、GCB+NH2柱、C18柱、PSA柱、弗羅里硅土柱、中性氧化鋁柱等,研究發(fā)現(xiàn),NH2柱(500 mg/3 mL)、GCB+NH2柱(500 mg/500 mg/6 mL)、中性氧化鋁柱(500 mg/3 mL)均會吸附內(nèi)標(biāo)4-羥基苯甲酸異丁酯。進(jìn)一步比較了C18柱(500 mg/3 mL)、PSA柱(500 mg/3 mL)、弗羅里硅土柱(500 mg/3 mL)對提取液的凈化效果,結(jié)果顯示,對于肝臟類樣品,PSA柱、弗羅里硅土柱可以吸附提取液中大部分極性色素,但是洗脫液濃縮復(fù)溶過膜后呈黃色渾濁狀,表明這2種固相萃取柱對其他雜質(zhì)的凈化效果不明顯,與李南等[7]的研究結(jié)果相近。而提取液經(jīng)C18柱凈化、濃縮、復(fù)溶過膜后溶液較為澄清透明,當(dāng)C18固相萃取柱規(guī)格提高至2 g/12 mL時(shí),復(fù)溶液過膜后明顯變得澄清透明。因此,實(shí)驗(yàn)最終選擇C18固相萃取柱(2 g/12 mL)進(jìn)行凈化。
2.4 方法學(xué)評價(jià)
2.4.1 基質(zhì)效應(yīng)
基質(zhì)效應(yīng)是影響結(jié)果準(zhǔn)確性的一個(gè)重要因素,在質(zhì)譜方法開發(fā)和確證過程中需要對基質(zhì)效應(yīng)做出評價(jià)。為了考察本方法在去除基質(zhì)效應(yīng)方面的效果,按照MATUSZEWSKI等[15]的方法對其進(jìn)行基質(zhì)效應(yīng)評估。實(shí)驗(yàn)選擇豬肉、牛肝、雞蛋、牛奶4種陰性樣品,按照1.2節(jié)進(jìn)行前處理,獲得的基質(zhì)提取液分別配制質(zhì)量濃度為100 μg/L基質(zhì)標(biāo)準(zhǔn)溶液(含內(nèi)標(biāo)2 μg/L)。此外,用乙腈-水溶液[V(乙腈)∶V(水)=3∶2]配制同樣質(zhì)量濃度的標(biāo)準(zhǔn)溶液,按照公式(1)計(jì)算方法的基質(zhì)效應(yīng)。

(1)
研究表明,當(dāng)用外標(biāo)法計(jì)算時(shí),這4種基質(zhì)的基質(zhì)效應(yīng)值為92.5%~97.1%;當(dāng)用內(nèi)標(biāo)法計(jì)算時(shí),這4種基質(zhì)的基質(zhì)效應(yīng)值為94.3%~98.7%,說明采用該方法測定動物源性食品中噻節(jié)因殘留量時(shí)無需配制基質(zhì)標(biāo)準(zhǔn)曲線,主要是APCI對基質(zhì)效應(yīng)的敏感程度要低于ESI,與文獻(xiàn)[13-14]中方法相比,對雞肉、雞蛋中噻節(jié)因殘留的研究基質(zhì)干擾更小。
2.4.2 線性范圍和定量限
配制質(zhì)量濃度為1、2、5、10、50、100、200、500 μg/L的系列標(biāo)準(zhǔn)溶液(含內(nèi)標(biāo)2 μg/L),對噻節(jié)因和內(nèi)標(biāo)4-羥基苯甲酸異丁酯定量離子的峰面積之比(y)與噻節(jié)因的質(zhì)量濃度(x,μg/L)進(jìn)行線性回歸,繪制標(biāo)準(zhǔn)曲線,其線性方程為y=0.077 87x-0.024 62,相關(guān)系數(shù)(R2)為0.999 3。研究表明,噻節(jié)因在1~500 μg/L范圍內(nèi)線性關(guān)系良好,且滿足GB/T 27404—2008《實(shí)驗(yàn)室質(zhì)量控制規(guī)范 食品理化檢測》附錄F中對于確證方法相關(guān)系數(shù)≥0.99的要求。
在陰性樣品中添加噻節(jié)因,按照1.2節(jié)進(jìn)行前處理上機(jī)測定,以10倍信噪比(S/N)為定量限,得到噻節(jié)因的方法定量限為1.0 μg/kg,完全滿足各國限量標(biāo)準(zhǔn)要求,且同現(xiàn)有方法相比具有更高的靈敏度[3,12-14]。
2.4.3 準(zhǔn)確度和精密度
通過對陰性樣品進(jìn)行加標(biāo)回收,考察方法的準(zhǔn)確度和精密度。按照1.2節(jié)提取和凈化步驟,在豬肉、豬肝、牛肉、牛肝、羊肉、羊肝、雞肉、雞胗、雞蛋和牛奶10種基質(zhì)樣品中添加定量的噻節(jié)因標(biāo)準(zhǔn)溶液進(jìn)行加標(biāo)回收試驗(yàn),加標(biāo)量分別為1.0、10.0和100.0 μg/kg,每個(gè)加標(biāo)水平做6個(gè)平行,試驗(yàn)結(jié)果見表2。
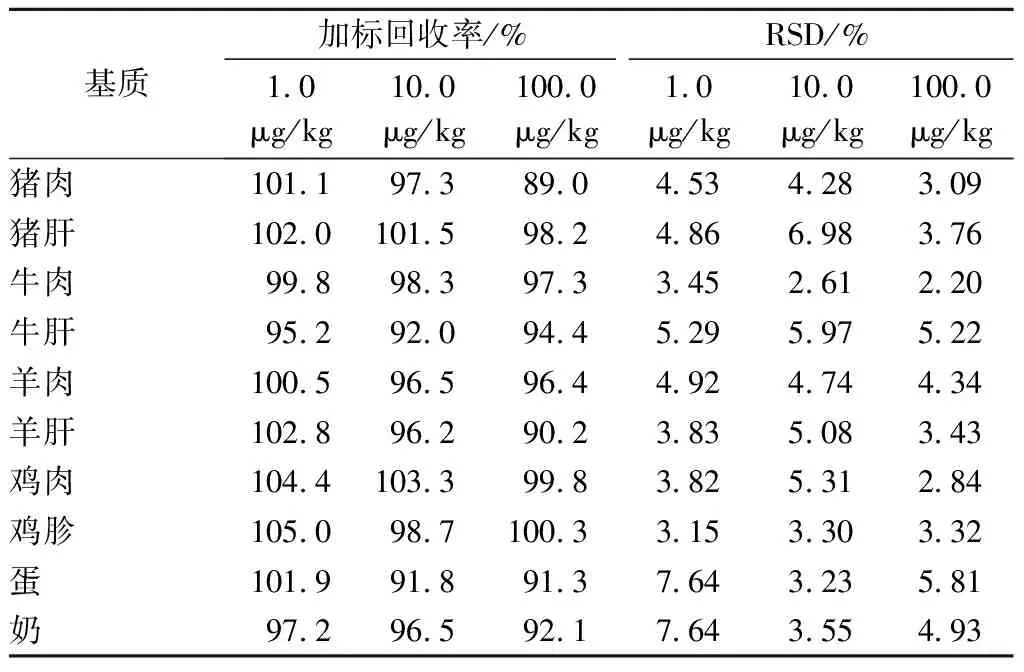
表2 不同基質(zhì)中噻節(jié)因的加標(biāo)回收率和精密度(n=6)
結(jié)果顯示,在3個(gè)加標(biāo)水平下,豬肉、豬肝、牛肉、牛肝、羊肉、羊肝、雞肉、雞胗、雞蛋和牛奶中噻節(jié)因的平均回收率為89.0%~105.0%,相對標(biāo)準(zhǔn)偏差為2.20%~7.64%。說明方法的準(zhǔn)確度高,通用性好,且方法的回收率優(yōu)于現(xiàn)有研究[3,12-14],完全滿足GB/T 27404—2008《實(shí)驗(yàn)室質(zhì)量控制規(guī)范 食品理化檢測》附錄F中檢測方法確認(rèn)回收率和精密度要求。
2.5 實(shí)際樣品的測定
運(yùn)用本文建立的方法,對市售的畜禽肉及肝臟類、蛋類、牛奶共40份樣品進(jìn)行噻節(jié)因的殘留檢測,所有樣品均為陰性。
3 結(jié)論
本文建立了高效液相色譜-串聯(lián)質(zhì)譜測定動物源性食品中噻節(jié)因殘留的分析方法,方法操作簡單,基質(zhì)干擾小,定性準(zhǔn)確,靈敏度高,線性關(guān)系、準(zhǔn)確度和精密度均滿足方法學(xué)指標(biāo),方法定量限完全滿足各國限量標(biāo)準(zhǔn)要求,可為動物源性食品中噻節(jié)因的殘留檢測提供技術(shù)手段。
