雙酶解-液液萃取-氣相色譜質譜法測定糕點中丙酸及其鹽類
2021-08-02 12:06:58王宇鐘玉心陳悅銘黃景初陳穗蘇燕瑜陳嘉欣蔡偉誼
食品與發酵工業 2021年14期
王宇,鐘玉心,陳悅銘,黃景初,陳穗,蘇燕瑜,陳嘉欣,蔡偉誼*
1(廣州市食品檢驗所,廣東 廣州,511400) 2(廣東美味鮮調味食品有限公司,廣東 中山,528437)
丙酸(propionic acid)及其鹽類[1-2]具有抑制微生物增殖和殺滅微生物的作用,常以丙酸鈣或丙酸鈉的形式用于食品防腐和防霉。糕點是富含蛋白質、淀粉、脂肪與糖類的食品[3],丙酸鈣在糕點制作過程中易與原料均勻混合,使用尤其普遍[4]。然而丙酸攝入過量,易患肥胖癥和糖尿病,食品安全國家標準GB 2760—2014規定糕點中丙酸及其鹽類使用限量不超過2.5 g/kg(以丙酸計)[5]。食安通食品抽檢與信息分析數據顯示,近5年糕點中丙酸及其鹽類超標導致不合格的事件時有發生。
食品中丙酸及其鹽類測定的樣品前處理主要采用直接浸提[6]、蒸餾提取[7]、液液萃取(溶劑提取)[8]、固相萃取[9]、沉淀法[10]等方法。糕點中蛋白質、脂肪、淀粉、糖類的含量較高,單一的液液萃取或者固相萃取無法完全去除蛋白質與脂肪。沉淀法去蛋白難以排除脂肪和其他添加劑(色素、甜味劑)的干擾,酸化、水蒸氣蒸餾法對蒸餾儀器要求高,且水溶液直接進樣容易造成氣相色譜襯管過載或色譜柱損壞。目前,針對丙酸的測定方法主要有離子色譜法[11]、高效液相色譜法[12-13]、氣相色譜法[14-16]、氣相色譜串聯質譜法(GC-MS)[17-18]。離子色譜法前處理過程復雜,測定中易損失目標物。高效液相色譜法需利用214 nm末端吸收檢測,容易受基質干擾,靈敏度不高。氣相色譜法具有較高靈敏度,但是定性分析與抗干擾能力較弱。因此,開發一種針對糕點中丙酸及其鹽類的簡便、準確的檢測方法具有重要意義。酶解法[19-20]在應對蛋白類或淀粉類樣品中有廣泛應用,液液萃取是傳統的前處理手段,通過調節pH值控制易電離化合物的化學形態,可以有效控制目標物的相間轉移,同時去除基質干擾(添加劑、糖類等極性雜質)。GC-MS具有全掃描與選擇離子監測工作模式,具有較好的定性與定量分析能力[21-22]。本研究采用淀粉酶-蛋白酶雙酶解樣品,乙醚去除脂肪等非極性雜質,調節pH值控制丙酸形態轉換并結合液液萃取、凈化,建立抗干擾強、高靈敏度的GC-MS檢測方法,滿足當前糕點中丙酸及其鹽類的檢測需求,為食品中添加劑殘留監管提供技術支撐。
1 材料與方法
1.1 材料與試劑
丙酸(純度99.5%),德國Dr.Ehrenstorfer公司;木瓜蛋白酶(酶活力≥2 400 U/g),北京百靈威技術有限公司;乙酸乙酯、乙醚、石油醚、正己烷(色譜純),美國Fisher;磷酸、硫酸、鹽酸、甲酸、乙酸(分析純)、α-淀粉酶(酶活力≥3 700 U/g),國藥集團化學試劑有限公司;磷酸氫二鈉、氯化鈉、氫氧化鈉(分析純),廣州化學試劑廠;實驗用水(一級水),美國Milli-Q超純水系統制備(18.2 MΩ/cm);實驗用糕點,市售。
1.2 儀器與設備
7890A-5975C氣相色譜-質譜聯用儀,美國Agilent公司;UW6200H分析天平,日本Shimadzu公司;CP225D分析天平,德國Sartorius公司;S470-B多功能pH 測試儀,瑞士METTLER TOLEDO公司;SW22恒溫水浴裝置,德國Julabo公司;MS3 digital旋渦混合器,德國IKA公司;Multi Reax渦旋振蕩器,德國Heidolph公司;Allegra X-30R離心機,美國BECKMAN公司;Milli-Q超純水系統,美國Millipore 公司。
1.3 實驗方法
1.3.1 標樣配制
準確稱取10 mg丙酸標準品,采用乙酸乙酯溶解并定容至10 mL容量瓶中,制成丙酸標準儲備液,于4 ℃ 冷藏保存,準確移取1 mL丙酸標準儲備液(1 mg/mL)于10 mL容量瓶中,乙酸乙酯定容,制成丙酸標準使用液(100 μg/mL),于4 ℃冷藏保存,備用。
分別移取0.01、0.05、0.5、1和2 mL的丙酸標準使用液(100 μg/mL)于5個10 mL容量瓶中,乙酸乙酯定容、搖勻,得0.10、0.50、5.0、10和20 μg/mL的標準曲線溶液。
1.3.2 樣品制備
取代表性樣品約500 g,裝入潔凈容器并密封,于4 ℃冷藏保存。
1.3.3 酶解
稱取2.50 g樣品于50 mL離心管,加入15 mL磷酸氫二鈉-磷酸緩沖溶液[50 mmol/L,pH值為(6.0±0.2)]溶解樣品,加入1.0 mL混合酶溶液(終濃度為質量分數5%的α-淀粉酶和木瓜蛋白酶),旋渦混合5 min,37 ℃恒溫水浴振搖40 min,得到樣品酶解液。
1.3.4 樣品的提取與凈化
樣品酶解液中加入0.6 mL 0.4 mol/L NaOH溶液,振搖30 min,加入1.0 g NaCl及20.0 mL乙醚,迅速旋渦混合5 min,搖勻后靜置分層,8 000 r/min離心2 min,去上層乙醚,保留下層溶液。
下層溶液中加入0.4 mL磷酸(使溶液pH<4),振搖2 min,得酸化溶液。
上述酸化溶液中加入10 mL乙酸乙酯,振搖2 min,8 000 r/min離心2 min,收集有機相至容量瓶中,殘渣加入10 mL乙酸乙酯,重復提取1次,合并2次萃取液至25 mL容量瓶中,加入乙酸乙酯定容,經0.22 μm濾膜過濾,氣相色譜質譜系統測定。
1.3.5 氣相色譜串聯質譜條件
(1)色譜條件:DB-WAX毛細管色譜柱(30 m×0.25 mm,0.25 μm);載氣及流速:氦氣(純度99.999%),流速1.0 mL/min;進樣口溫度220 ℃;程序升溫:初始溫度80 ℃,保持2 min,以10 ℃/min 升至200 ℃后,保持1 min,再以20 ℃/min 升至220 ℃,保持0 min,共16 min;分流進樣(40∶1),進樣量1 μL。
(2)質譜條件:離子源類型:電子轟擊源(EI);掃描方式:選擇離子模式(SIM);電離方式:EI;電離能量:70 eV;離子源溫度(TEM):230 ℃;MS四級桿溫度150 ℃;溶劑延遲 5 min。
(3)離子選擇參數:質譜定性分析離子為m/z45、55、57、73,定量分析離子為m/z74。
1.3.6 數據處理
將GC-MS中得到的化合物的質譜圖與美國國家標準與技術研究院(National Institute of Standards and Technology,NIST)數據庫進行對比,將匹配度>80%的化合物作為暫定結果,然后采用NIST MS Search 2.0 標準譜庫相匹配檢索定性,當正反匹配度均>800(最大值為1 000)時,確定化合物的類型,通過外標法計算丙酸含量。
采用Excel 2016進行數據整理、分析,并繪制圖表。
2 結果與分析
2.1 樣品前處理方法的優化與建立
2.1.1 酶解時間的優化
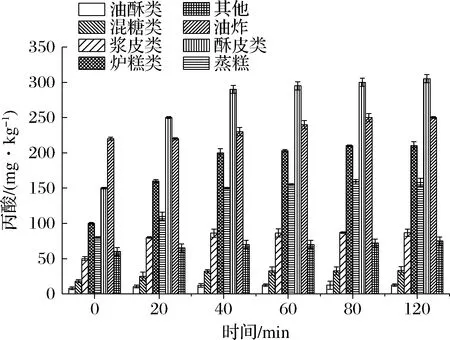
樣品中的淀粉含有大量的羥基、蛋白表面電荷會對丙酸產生靜電吸附,導致丙酸第一步堿化萃取時萃取效果下降,直接影響最終濃度。因此,本實驗中酶解效果以丙酸測定濃度進行判斷(所有樣品經檢測均為陽性)。結果表明,在0~120 min內不同類型糕點中檢測到丙酸的濃度隨著酶解時間的延長有不同程度的增加;當酶解時間為40 min時,丙酸的檢測濃度趨于穩定(圖1),推斷淀粉、蛋白質已酶解完全。本實驗優化后的萃取時間為40 min。
2.1.2 pH值的優化
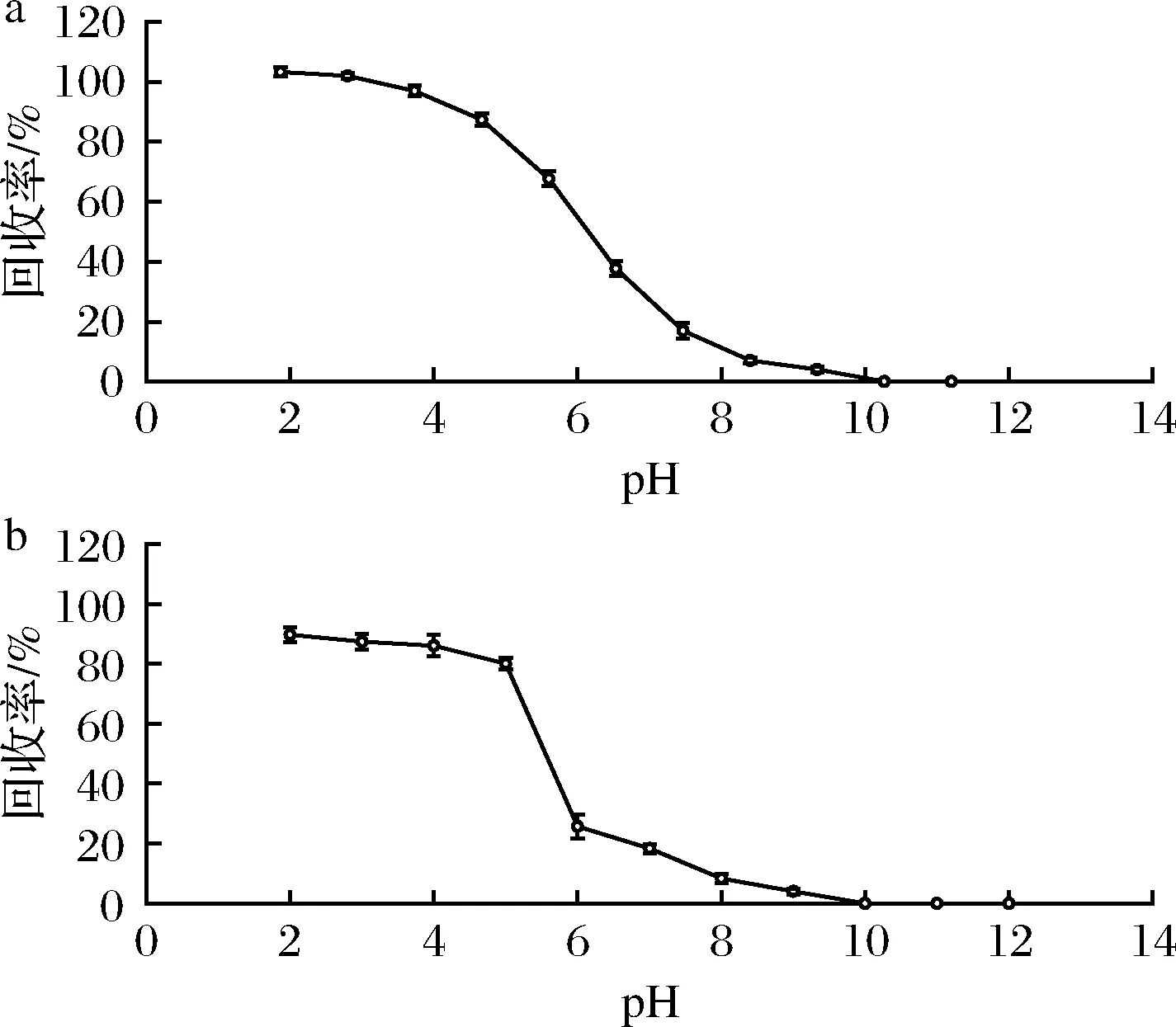
由于丙酸為弱酸性,丙酸在水溶液中的解離平衡常數(pKa)為5.001 9[23],強堿時極不穩定,油/水分配系數隨pH的增大而降低。本文通過提高水溶液 pH 值,使丙酸成鹽而降低溶解性,達到去除脂肪的目的,再降低水溶液 pH 值,增加丙酸在乙酸乙酯的溶解性,并減少水溶性雜質引入,達到提取丙酸的目的。當pH>pKa+2=7時,形成丙酸解離狀態,隨著pH的降低,當pKa-2
2.1.3 脫脂溶劑種類和用量的選擇
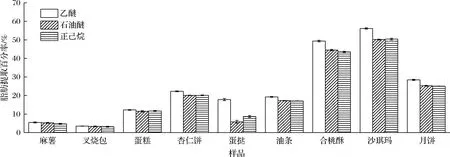
本文采取的是溶劑脫脂法,考察選取一種與脂肪性質相似溶劑浸入酶解的水溶液中以除去脂肪。試驗結果表明,當糕點脂肪提取百分率<15%,所選3種脫脂溶劑的脫脂效果基本相同。但是,當糕點脂肪提取百分率>15%,乙醚提取的脂肪含量較高,整體有機溶劑脫脂效果為乙醚>石油醚=正己烷。尤其是含磷脂高或膠原蛋白高的樣品(如蛋撻),乙醚提取脂肪的效果優于石油醚或正己烷。主要原因可能是石油醚與正己烷的極性指數相同(P=0.1),乙醚的極性指數較大(P=2.8),只有極性較大的乙醚能夠滲透入含有膠原蛋白較多的乳化水溶液中提取脂肪,也只有乙醚能較好溶解極性磷脂等物質(圖3)。
在對不同脂肪含量糕點的脂肪提取試驗中,脂肪提取百分率隨著乙醚量增加而升高,當乙醚的用量≥20 mL時,不同脂肪含量糕點的脂肪提取百分率基本趨于一致,接近糕點中脂肪的含量,說明試驗使用20 mL 乙醚可以對不同脂肪含量的糕點達到脫脂效果(圖4)。

2.1.4 酸化試劑的選擇
調節溶液的pH值<4.0,使丙酸處于非電離狀態,在有機相液液萃取時能完全進入到有機相中。甲酸和乙酸的pKa分別為3.74、4.74,調節溶液pH時用量約為2.0 mL,磷酸的pKa為1.52、鹽酸、硝酸、硫酸的pKa均<1,用量均為0.4 mL。結果如圖5所示,甲酸、乙酸等有機酸會干擾丙酸的測定,推斷其原因可能是:甲酸、乙酸與丙酸結構相似,存在加減整數倍CH2質量分數(14),且在選擇離子模式狀態下,分子離子容易斷裂產生相似的碎片離子從而干擾丙酸的定量;甲酸和乙酸的加入量較大,有一部分會以分子形態進入到有機相中,且屬于易揮發酸,會干擾到目標物的檢測。磷酸、鹽酸、硝酸、硫酸等無機酸干擾較小,4種無機酸的酸化效果相差不大。考慮到磷酸的無強氧化性、無強腐蝕性、安全性和弱干擾性,試驗選擇磷酸為酸化試劑。

2.2 色譜柱的選擇和色譜質譜條件優化
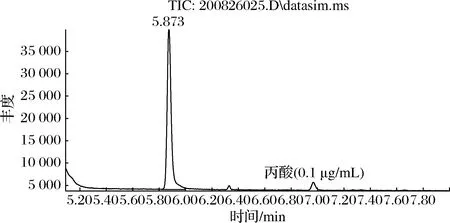
丙酸屬于極性物質,試驗考察了HP-INNOWAX、DB-WAX、DS-FFAP色譜柱,結果表明DB-WAX色譜柱分離效果最好,丙酸峰型好,保留時間為7.00 min(圖6)。采用全掃描模式對適宜濃度的標準溶液進行掃描,得到丙酸相對應的母離子保留時間,對其母離子進行碎片離子掃描,選擇豐度較高的離子作為定量離子,另外4組作為定性離子[26],通過對色譜、質譜條件優化,選擇丙酸質譜定性分析離子為m/z45、55、57、73,定量離子為m/z74(圖7)。

2.3 線性范圍、檢出限和定量限
按照1.3.5方法進行進樣,按照1.3.1方法配制0.10、0.50、5.0、10和20 μg/mL標準曲線。選擇已經確證的陰性樣品加入丙酸標準品,計算3倍信噪比(S/N)所對應的樣品質量濃度確定方法檢出限。結果顯示在0.100~20.0 μg/mL內濃度(y)與峰面積(x)的線性關系良好,對應線性方程為y=2 460x+302,相關系數r2=1.000,檢出限0.450 mg/kg。
2.4 回收率和精密度
選取8種類別的糕點樣品(扣除本底值),在低(1.25 mg/kg)、中(12.5 mg/kg)、高(125 mg/kg)3個水平進行加標回收試驗(n=6),并考察6次測定結果的相對標準偏差。結果如表1所示,平均回收率在83%~109%,相對標準偏差在1.76%~6.24%,表明方法準確可靠。
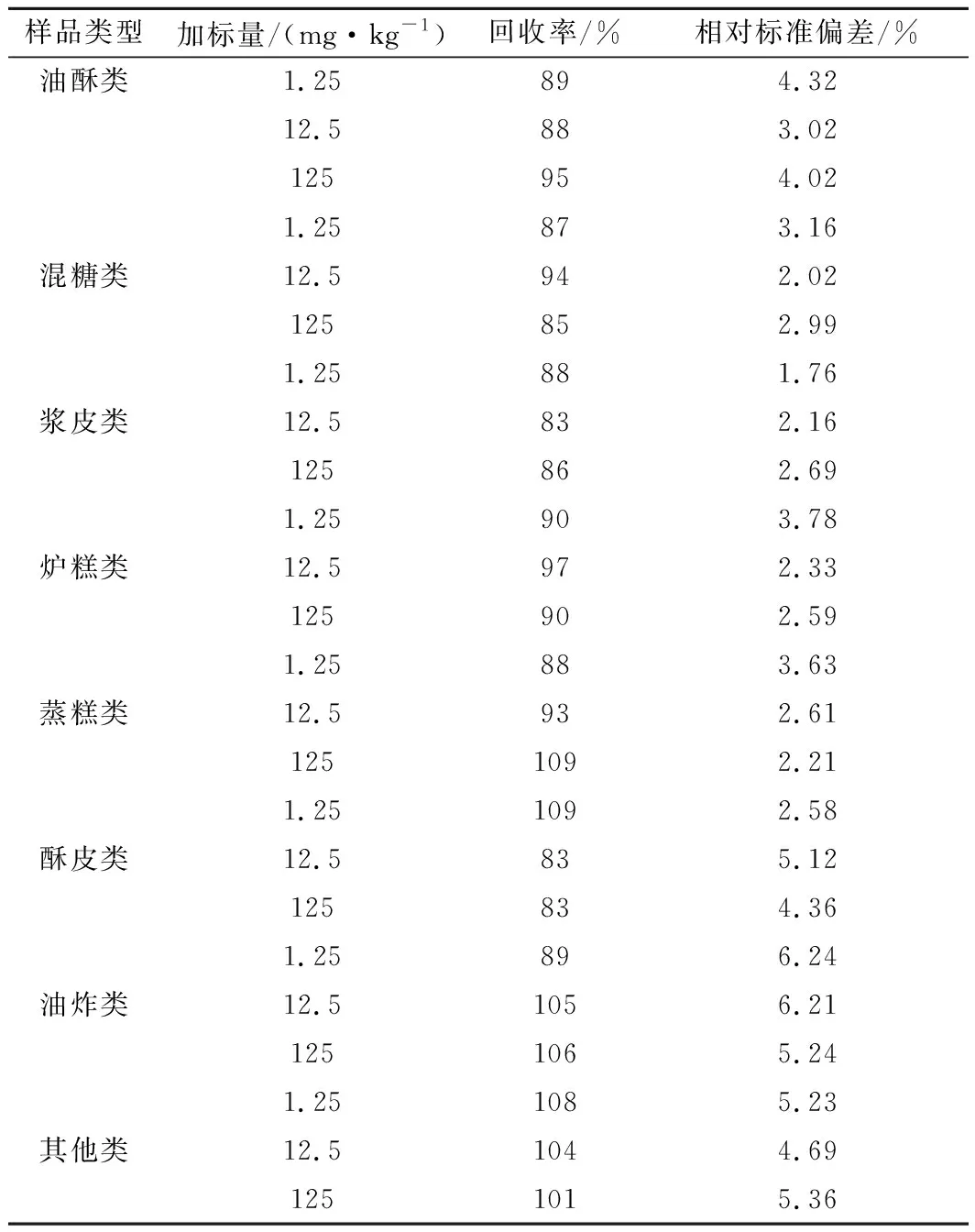
表1 加標回收率及精密度測試結果(n=6)Table 1 Recovery and precision test results (n=6)
2.5 實際樣品的測定
抽取廣州市不同區域市售糕點樣品,采用本文所建立的方法進行測定。結果表明所測市售糕點樣品的丙酸的含量在12.3~1 700 mg/kg(表2),均符合國家標準限量要求。
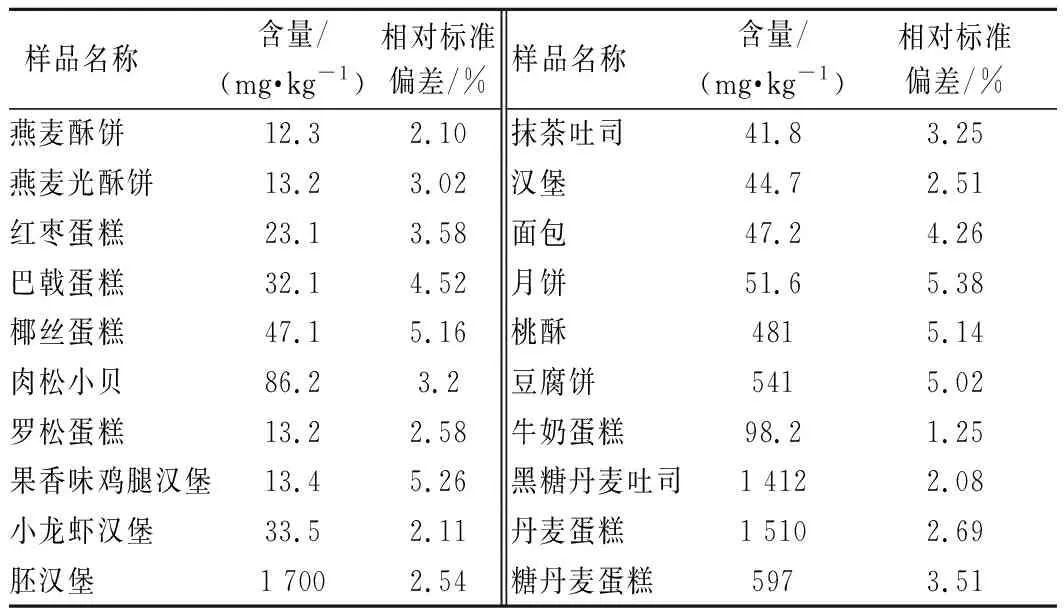
表2 實際樣品測試結果(n=3)Table 2 The detection results of real sample (n=3)
3 結論
目前,關于糕點中丙酸的質譜檢測研究鮮有報道,針對糕點中丙酸檢測的前處理方法研究也較少。本試驗在前期研究基礎上成功建立了測定糕點中丙酸含量的雙酶解-液液萃取-氣相色譜質譜分析方法。與現行方法相比,本研究所建立的前處理方法能有效去除蛋白質、脂肪等基質對檢測信號的干擾,且操作簡便、時間短、效率高,分析方法靈敏度高、選擇性好、結果準確,能夠滿足糕點這種富含蛋白、脂肪樣品中的丙酸殘留檢測需求,為糕點中丙酸的測定和質量監控提供新的技術補充。
