通過式固相萃取與液相色譜串聯質譜和氣相色譜串聯質譜結合測定加熱卷煙中多種農藥殘留
2021-08-04 03:50:10楊飛紀元劉珊珊王穎范子彥鄧惠敏邊照陽陳曉水唐綱嶺
中國煙草學報 2021年3期
楊飛,紀元,劉珊珊*,王穎,范子彥,鄧惠敏,邊照陽,陳曉水,唐綱嶺
1 國家煙草質量監督檢驗中心,鄭州高新技術產業開發區翠竹街6號 450001;
2 浙江中煙工業有限責任公司技術中心,杭州市西湖區科海路118號 310024
近年來,加熱煙草制品(HTPs)在市場上的占有率逐年上升,而對于加熱煙草制品的研究也逐漸成為當前的熱點[1-2]。加熱煙草制品是利用特殊熱源對由煙絲或其它煙草材料制備的煙芯進行加熱(150℃~500℃),產生可吸入氣溶膠。加熱煙草制品因其無需高溫燃燒煙草,僅在相對低溫下對煙草原料進行加熱,減少了煙草高溫燃燒裂解產生的有害成分,但同時增加了某些農藥殘留的轉移風險[3-5]。
蔬菜、水果及煙草等農作物在種植和儲藏期間均會普遍施用農藥防治病蟲害,以提高產量并獲得高質量的產品。但是,農作物中的農藥殘留可能對人類健康產生諸多不利影響。由于農藥種類多,性質差異較大,且農藥殘留含量低,很難采用單一的分析方法對多農藥殘留進行檢測。目前,氣相色譜-串聯質譜(GC -MS/MS)[6-8]和液相色譜-串聯質譜(LC-MS/MS)[9-10]已經成為測定多種基質中的多種農藥殘留的“金標準”。串聯質譜(tandem mass spectrometry,MS/MS)由兩個或更多的質譜串接而成,最簡單的串聯質譜由兩個質譜串聯而成。第一級質譜使試樣中各組分在離子源中發生電離,生成不同荷質比的母離子,母離子經過氮氣、氬氣等碰撞氣碰撞產生碎片離子,碎片離子經過第二級質譜進行質量分析。串聯質譜具有高靈敏度和高選擇性,是一種分析復雜基質中農藥殘留的有效手段。
煙草基質復雜,色素、脂質等含量較高,對樣品中的痕量農藥殘留進行分析前,需要進行前處理去除基質干擾以提高方法的靈敏度和特異性。現有分析方法多采用固相萃取(SPE)[6]和QuEChERS[7-10]技術凈化樣品。但這些方法前處理過程繁瑣,耗時長。PRiME(process,robustness,improvements,matrix effects, ease of use)HLB柱是一種新型親水親脂平衡萃取柱,其特有的親脂基團可以有效地吸附煙草中的色素和磷脂類物質。與傳統的固相萃取柱相比,PRiME HLB固相萃取柱是一種通過式固相萃取柱,不需要活化/平衡、清洗或洗脫步驟,樣品提取液依靠重力通過PRiME HLB柱并收集,雜質被吸附在固相萃取柱上。PRiME HLB通過式固相萃取具有簡單、高效、環保且易于操作的優勢,尤其適用于復雜樣品中分析物的高通量分析[11-15]。
本方法采用PRiME HLB通過式固相萃取凈化、液相色譜-串聯質譜和氣相色譜-串聯質譜同時檢測加熱卷煙中的殺菌劑、殺蟲劑和除草劑等53種常見的農藥殘留,旨在為加熱卷煙中多農殘的檢測提供了準確、便捷、快速的分析平臺。
1 材料與方法
1.1 材料、試劑和儀器
25個不同品牌的加熱卷煙樣品全部采購于國外市場。所有樣品除去濾嘴和卷煙紙后,將煙絲粉碎,并過2 mm(10目)篩后貯存于棕色玻璃瓶中,4℃下保存。
22種農藥的混合標準溶液Ⅰ(100 μg/mL,上海安譜實驗科技股份有限公司);31種農藥的混合標準溶液Ⅱ(100 μg/mL,上海安譜實驗科技股份有限公司);內標d4-三唑酮和三苯基磷酸酯(1 mg,純度大于98.0%,德國Augsburg公司);乙腈、甲苯和甲酸(色譜純,德國Merck公司);無水硫酸鎂(MgSO4)、氯化鈉(NaCl)、乙二胺基-N-丙基(PSA)(分析純,上海百靈威科技股份有限公司);水為超純水;高純氮、高純氬、高純氦(純度> 99.999%,鄭州源正科技)。PRiME HLB通過式固相萃取柱(60 mg,3 cc,美國Waters公司)。
ACQUITY I class超高效液相色譜儀、Xevo-TQS四極桿串聯質譜儀(配電噴霧電離源,美國Waters公司);Agilent 7890B-5977A氣相色譜-三重四極桿質譜聯用儀(美國Agilent科技有限公司);ACQUITY UPLC BEH C18柱(100 mm×2.1 mm,1.7 μm,美國Waters公司);Turbo Vap氮吹儀(美國Biotage公司);VX200渦旋振蕩儀(美國Labnet公司);SG3-30K高速離心機(德國Sigma公司);Milli-Q超純水系統(美國Millipore公司);BSA2245-CW電子天平(感量:0.0001 g,德國Sartorius公司)。
1.2 方法
1.2.1 LC-MS/MS用基質匹配標準工作溶液
將1.0 mg內標d4-三唑酮用乙腈溶解并轉移至100 mL容量瓶中,用乙腈定容至刻度,制得內標工作液Ⅰ(10 μg/mL)。
移取1.0 mL的混合標準溶液Ⅰ于10 mL容量瓶中,用乙腈溶解并定容至刻度,制得的質量濃度為10 μg/mL的混合標準儲備液Ⅰ。
分別移取混合標準儲備液Ⅰ 500、200、100、50、20 μL至5個10 mL容量瓶中,并加入100 μL內標工作液Ⅰ,用超純水定容,制得質量濃度分別為500、200、100、50及20 ng/mL的標準工作溶液Ⅰ。
分別移取0.2 mL標準工作溶液Ⅰ和0.2 mL空白樣品提取液,混合后用超純水稀釋至1 mL,即得到基質匹配系列標準工作溶液Ⅰ。所有溶液避光儲存在4℃的冰箱中,使用前將其恢復至室溫。
1.2.2 GC-MS/MS用基質匹配標準工作溶液
將1.0 mg內標三苯基磷酸酯用甲苯溶解并轉移至50 mL容量瓶中,用甲苯定容至刻度,制得內標工作液Ⅰ(20 μg/mL)。
移取1.0 mL的混合標準溶液Ⅱ于10 mL容量瓶中,用甲苯定容至刻度,制得的質量濃度為10 μg/mL的混合標準儲備液Ⅱ。
分別移取混合標準儲備液Ⅱ 500、200、100、50、20 μL至5個10 mL容量瓶中,并加入200 μL內標工作液Ⅱ,用甲苯定容,制得質量濃度分別為500、200、100、50及20 ng/mL的標準工作溶液Ⅱ。
分別取5份空白提取液各1 mL,置于氮吹濃縮儀適用的試管中,在氮吹濃縮儀上濃縮近干,再分別加入1 mL不同濃度的標準工作溶液Ⅱ,攪拌復溶,即得到基質匹配系列標準工作溶液Ⅱ。
所有溶液避光儲存在4℃的冰箱中,使用前將其恢復至室溫。
1.2.3 煙絲樣品的提取和凈化
稱取2 g粉碎后的樣品(精確至0.01 g)于50 mL具蓋離心管中,加入10 mL水,振蕩,讓水充分浸潤樣品。移取10 mL乙腈至離心管中,并加入100 μL內標工作液Ⅰ和200 μL內標工作液Ⅱ,然后將離心管置于渦漩混合振蕩儀上以2000 r/min的速率振蕩2 min。然后向離心管中加入4 g無水硫酸鎂和1 g氯化鈉、1 g檸檬酸鈉和0.5 g檸檬酸二氫鈉,立即于漩渦混合振蕩儀上以2000 r/min振蕩2 min,然后以5000 r/min離心5 min。
移取2.0 mL的上清液到PRiME HLB柱中,且在重力作用下讓其自然通過小柱,收集濾液。濾液經0.22 μm有機相濾膜過濾后,移取200 μL用乙腈稀釋至1 mL進LC-MS/MS檢測。
在上述50 mL的離心管中繼續加入5 mL甲苯,渦旋混勻2 min,然后以5000 rpm離心5 min。移取2.0 mL的提取液到PRiME HLB柱中,且在重力作用下讓其自然通過小柱,并收集濾液。濾液經0.22 μm有機相濾膜過濾后進GC-MS/MS檢測。
1.2.4 空白樣品的提取
稱取2 g空白樣品于50 mL具蓋離心管中,加入10 mL水和10 mL乙腈至離心管中,然后將離心管置于渦漩混合振蕩儀上以2000 r/min的速率振蕩2 min。然后向離心管中加入4 g無水硫酸鎂和1 g氯化鈉、1 g檸檬酸鈉和0.5 g檸檬酸二氫鈉,立即于漩渦混合振蕩儀上以2000 r/min振蕩2 min,然后以5000 r/min離心5 min。
移取2.0 mL的上清液到PRiME HLB柱中,且在重力作用下讓其自然通過小柱,收集濾液。濾液經0.22 μm有機相濾膜過濾,該溶液用于制備LC-MS/MS用基質匹配標準工作溶液。
在上述50 mL的離心管中繼續加入5 mL甲苯,渦旋混勻2 min,然后以5000 rpm離心5 min。移取2.0 mL的提取液到PRiME HLB柱中,且在重力作用下讓其自然通過小柱,并收集濾液。濾液經0.22 μm有機相濾膜過濾,該溶液用于制備GC-MS/MS用基質匹配標準工作溶液。
1.3 實驗條件
1.3.1 LC-MS/MS分析條件
ACQUITY UPLC BEH C18柱(100 mm×2.1 mm,1.7 μm);柱溫:40℃;進樣量:2 μL;流速:0.3 mL/min;流動相:A相為乙腈,B相為0.1%(體積分數)甲酸水溶液;梯度洗脫程序:0~0.50 min為10% A,0.50~9.00 min為10% A~100% A,9.00~12.25 min為100% A,12.25~12.35 min為100% A~10% A,12.35~15.00 min為10% A。質譜掃描方式:正離子掃描;離子源:電噴霧電離源(ESI);離子源溫度:150℃;脫溶劑氣溫度:350℃;脫溶劑氣流量:800 L/h;錐孔氣流量:60 L/h;毛細管電壓:2.6 KV;采集模式:多反應監測(MRM);質譜參數見表1。內標法定量,內標為d4-三唑酮。
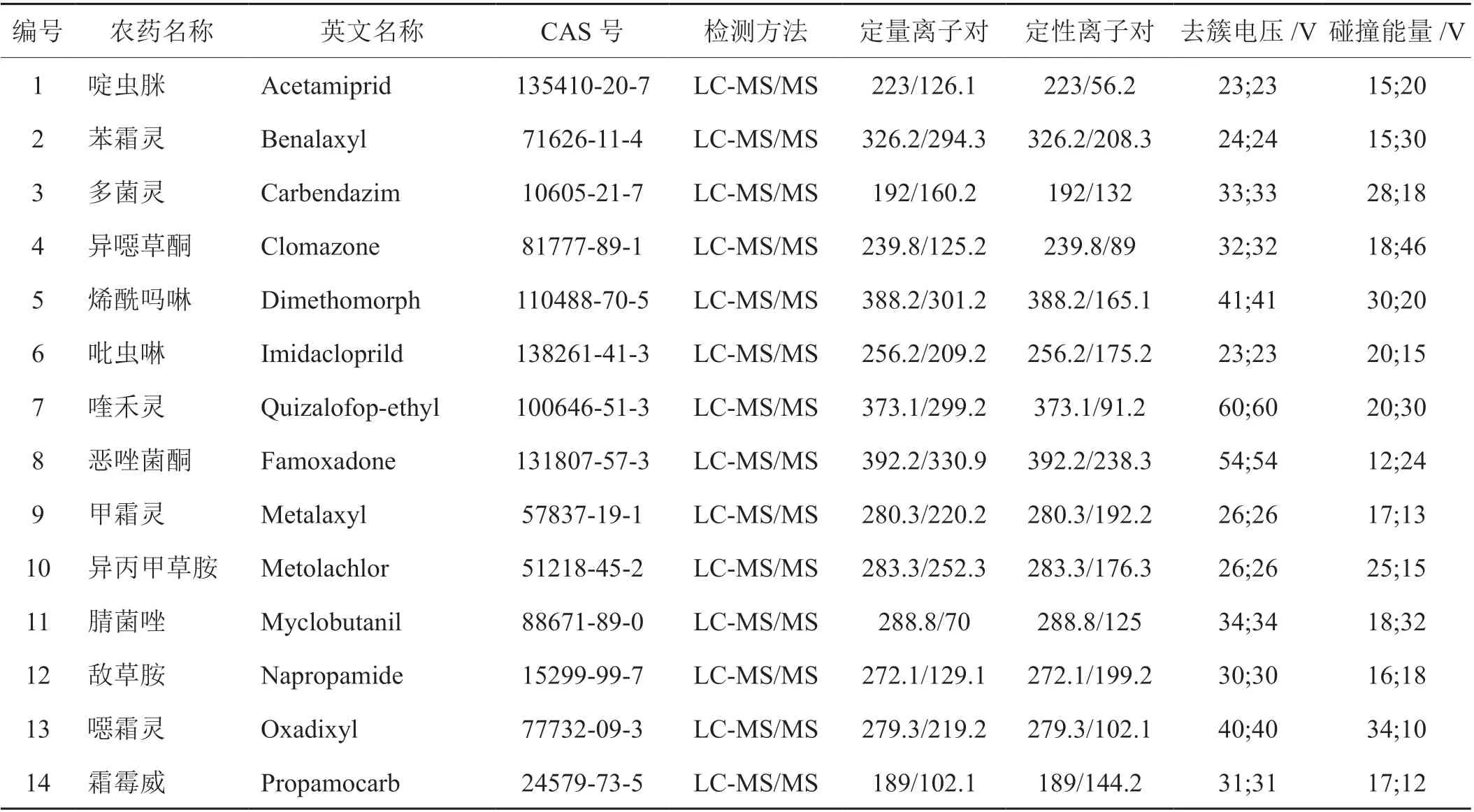
表1 分析物的液相色譜串聯質譜參數Tab. 1 LC-MS/MS parameters for analytes

續表1
1.3.2 GC-MS/MS分析條件
色譜柱:Agilent HP -5MS 毛細管色譜柱(30 m×0.25 mm×0.25 μm);PTV進樣口,初始溫度70℃,進樣后以4℃/s升至280℃;進樣量1 μL,不分流進樣;載氣:氦氣,恒流模式,流速為1.2 mL/min;柱溫箱升溫程序:初始溫度為90℃,初始時間為5 min;以25℃/min速率由90℃升至180℃,保持15 min;然后以5℃/min升至280℃,保持6.5 min;然后以40℃/min升至300℃,保持5 min;EI電離模式,電離能70 eV;離子源溫度250℃,傳輸線溫度280℃;碰撞氣:氮氣,0.16 MPa;溶劑延遲:5 min;采集模式:多反應監測(MRM);質譜參數見表2。內標法定量,內標為三苯基磷酸酯。

表2 分析物的氣相色譜-串聯質譜參數Tab. 2 GC-MS/MS parameters for analytes
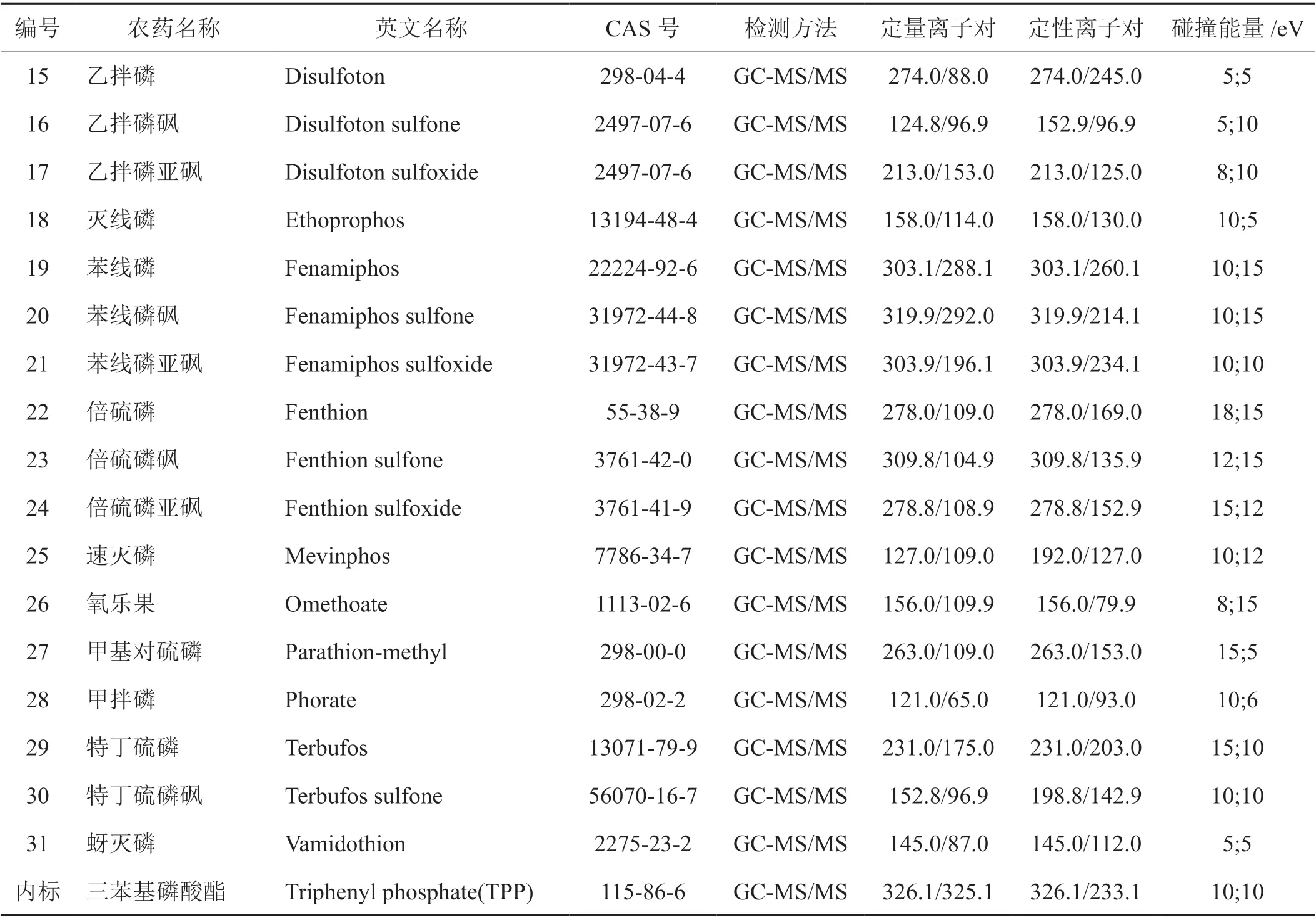
續表2
2 結果與討論
2.1 凈化效果的比對
為驗證方法優越性,將PRiME通過式固相萃取的凈化效果與PSA分散固相萃取的凈化效果[11-12]進行了比較,以基質效應(ME)的大小進行表征。其中,基質效應的計算公式為ME=(基質匹配標準溶液曲線斜率/純溶劑標準溶液曲線斜率-1)× 100%。ME絕對值越大表明基質效應越強[13]。部分農藥基質效應見表3。結果顯示,經PRiME HLB柱凈化后,大部分農藥的基質效應與PSA分散固相萃取方法相當,個別農藥的基質效應明顯低于PSA分散固相萃取方法。且經PRiME凈化后的提取液較PSA分散固相萃取凈化后的提取液顏色更淺(如圖1)。因此本研究采用PRiME HLB通過式固相萃取柱凈化樣品。該方法僅需樣液上柱,無需活化、淋洗、洗脫等步驟,操作更快捷方便,且PRiME HLB固相萃取柱填料因鍵合特有親脂基團,能有效吸附煙草基質中的磷脂、脂肪、色素等干擾物,從而實現樣品凈化目的。樣品經PRiME HLB固相萃取和PSA分散固相萃取后部分農藥的選擇離子色譜圖如圖2所示。
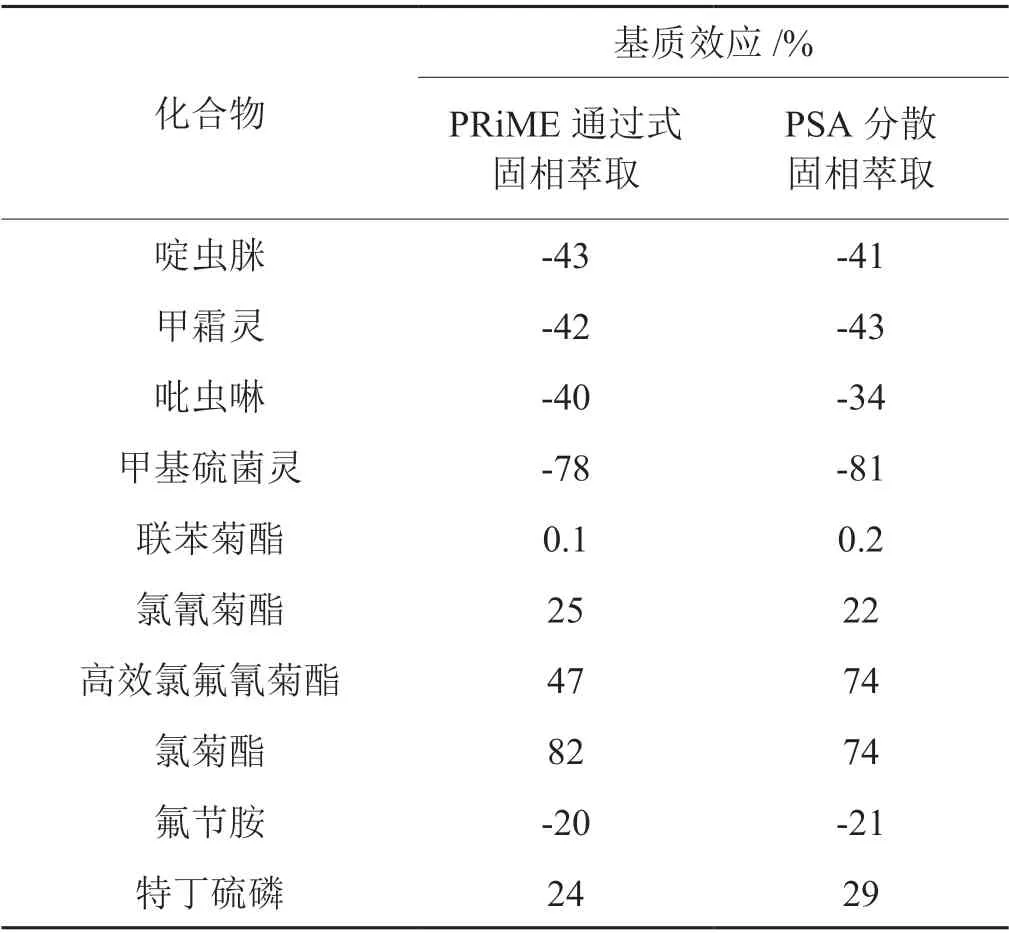
表3 不同凈化方法基質效應的對比Tab. 3 Comparison of matrix effects of different purification methods
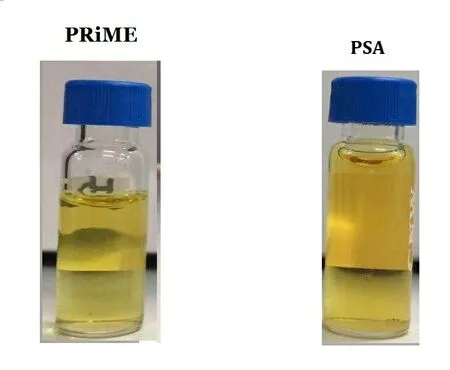
圖1 不同凈化方法凈化效果的對比Fig.1 Comparison of purification effects of different purification methods
2.2 方法對比
為了驗證PRiME通過式固相萃取方法的準確性,我們采用YC/T 405.1-2011《煙草及煙草制品 多種農藥殘留量的測定 第1部分:高效液相色譜-串聯質譜法》、YC/T 405.2-2011《煙草及煙草制品 多種農藥殘留量的測定 第2部分:氣相色譜-串聯質譜法》和本方法進行了對比實驗。煙草行業標準YC/T 405.1-2011、YC/T 405.2-2011采用QuEChERS法對樣品進行提取后、使用PSA進行分散固相萃取凈化,然后以LC-MS/MS或GC-MS/MS檢測。采用本方法與煙草行業標準對2019年和2020年Fapas國際能力驗證的4個樣品進行檢測,結果(表4)顯示,本方法檢測結果與標準方法檢測結果之間無明顯差異,說明兩種方法的一致性較好。

圖2 100 ng/mL的標準溶液中部分農藥的選擇離子色譜圖(A)PRiME通過式固相萃取(B)PSA分散固相萃取Fig.2 MRM chromatograms of some pesticides in 100 ng/mL standard solution (A) PRiME pass-through solid phase extraction (B) PSA dispersive solid phase extraction

表4 不同方法的測定結果對比Tab. 4 Comparison of testing results of different methods
2.3 方法評價
2.3.1 基質效應
由于加熱卷煙的煙芯基本上都是采用的再造煙葉,國內外再造煙葉的原料的不同,因此本文分別考察了烤煙、白肋煙和晾曬煙中部分農藥的基質效應。如表5所示,GC-MS/MS分析時,基質對大部分農藥有顯著的增強效應,LC-MS/MS分析時,基質對大部分農藥有顯著的抑制效應,因此定量分析均需采用基質匹配標準工作曲線。

表5 不同種類樣品基質效應的對比Tab. 5 Comparison of matrix effects of different types of samples
2.3.2 標準曲線、檢出限和定量限
在優化的色譜、質譜條件下,對配制好的基質匹配系列標準工作溶液Ⅰ和基質匹配系列標準工作溶液Ⅱ分別進行測定,以目標物峰面積和內標的峰面積比為縱坐標(Y),對應的質量濃度值為橫坐標(X,ng/mL),繪制標準工作曲線,其線性方程及線性相關系數(r)見表6。可以看出,在20~500 ng/mL 濃度范圍內,均能呈現良好的線性關系,其線性相關系數均大于0.9977。將加標空白基質中產生的信噪比(S/N)等于3的濃度定義為檢出限(LOD),信噪比(S/N)等于10時的濃度為定量限(LOQ)。如表6所示,LOD和LOQ范圍分別在0.31~10.88 μg/kg和1.11~36.21 μg/kg之間。
2.3.3 方法回收率及精密度
采用加標回收實驗來測試方法的準確度和精密度,該方法的精密度由重復性實驗確定,并以相對標準偏差(RSD)表示。在空白樣品中加入總量濃度為0.05、1.0、2.0 mg/kg的混合農藥,制備得到3個不同濃度的加標樣品,按照上述方法對加標樣品進行處理,每個濃度的樣品重復測定5次。結果(表6)顯示,在所有濃度水平下均獲得令人滿意的平均回收率(77.36%~108.28%),相對標準偏差(RSD)為1.4%~7.6%。表明該方法準確度高,重復性好,可滿足加熱卷煙中多種農藥殘留量的檢測需求。
2.4 實際樣品的檢測
應用所建立的方法對國外生產的25個不同品牌的加熱卷煙樣品進行檢測,所有樣品檢出殺菌劑,主要是霜霉威和甲霜靈,個別樣品檢出有多菌靈。其中霜霉威的檢出值為0.09~0.45 mg/kg。另外,19批次樣品檢出殺蟲劑吡蟲啉,檢出值為0.10~0.22 mg/kg。有機磷和擬除蟲菊酯類農藥未見檢出。所有檢出值均遠低于Coresta制定的農用化學品指導性殘留限量。

表6 線性系數,檢出限,定量限和精密度Tab. 6 Correlation coefficient, LODs, LOQs and precisions
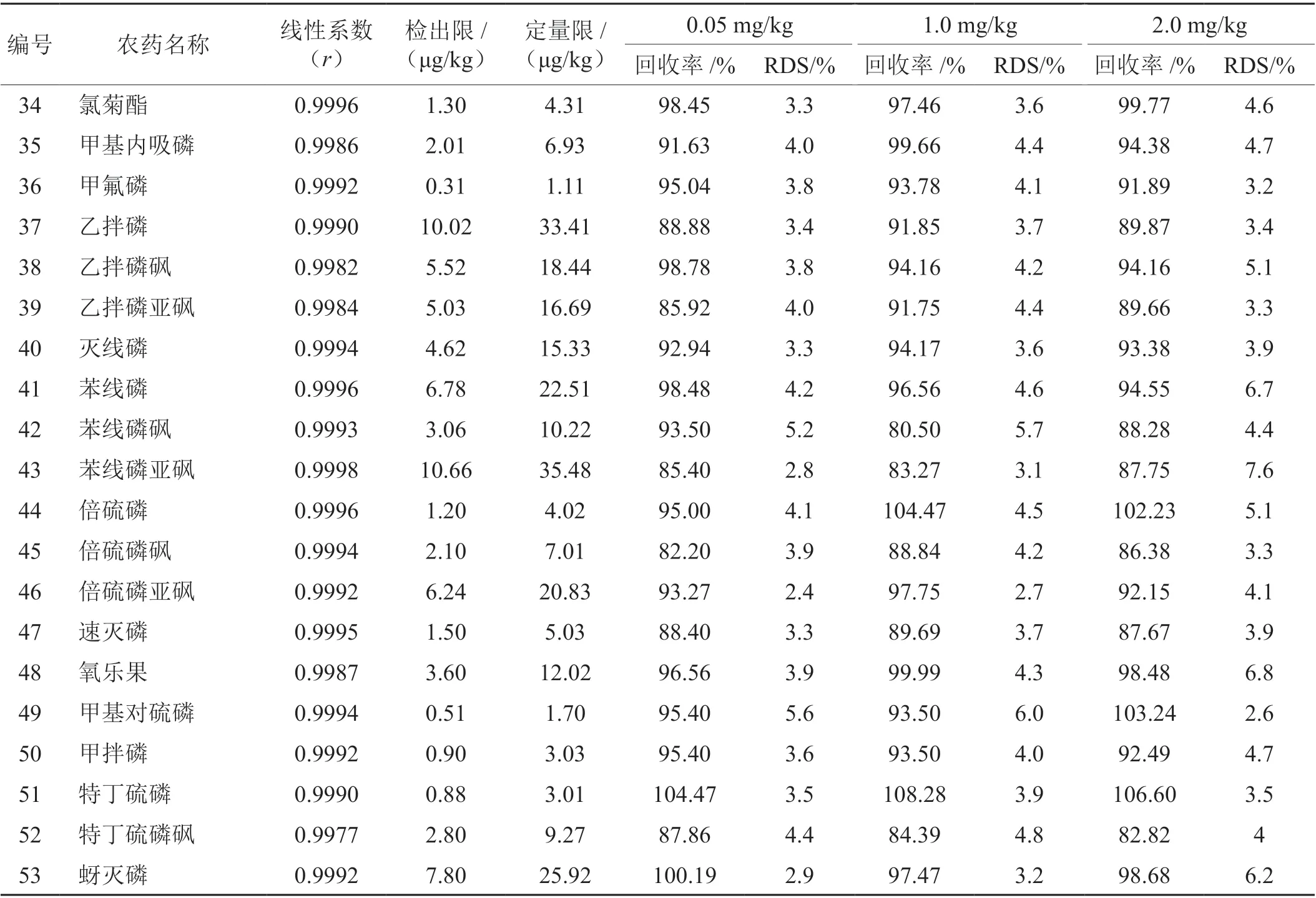
續表6
3 結論
通過優化質譜條件、色譜條件及樣品前處理過程等,建立了LC-MS/MS和GC-MS/MS法結合PRiME HLB通過式固相萃取測定加熱卷煙中53種農藥殘留的方法。在優化后的實驗條件下,53種農藥的回收率在77.36%~108.28%之間,相對標準偏差在1.4%~7.6%之間。國際能力驗證實驗結果表明,該方法可以對盲樣進行準確的定性和定量分析。該方法定量結果準確可靠,前處理簡單快速、時間短,能滿足加熱卷煙中多種農藥殘留分析的要求,可用于加熱卷煙中農藥殘留的實際檢測。
猜你喜歡
城市道橋與防洪(2022年4期)2022-07-01 06:04:12
兒童故事畫報(2019年5期)2019-05-26 14:26:14
當代陜西(2019年8期)2019-05-09 02:22:48
動漫星空(興趣百科)(2019年3期)2019-03-07 07:23:10
家庭影院技術(2018年4期)2018-05-09 07:07:52
Coco薇(2016年2期)2016-03-22 02:42:52
專用汽車(2016年4期)2016-03-01 04:13:43
質量與標準化(2015年9期)2015-12-31 11:41:40
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年7期)2015-08-11 15:03:12
