膠質纖維酸性蛋白星型細胞病的病例特點
2021-08-05 05:37:06譚淑慧
中風與神經疾病雜志 2021年6期
關鍵詞:信號
袁 泉, 譚淑慧
自身免疫性膠質纖維酸性蛋白星型細胞病是一種以膠質纖維酸性蛋白(glial fibrillary acidic protein,GFAP)抗體為標志物的神經系統自身免疫性疾病,其特異性IgG抗體已經在患者和動物身上被證實,這種抗體選擇性的作用于星型膠質細胞的膠質纖維酸性蛋白,于2016年被首次報道[1,2]。其好發于40歲以上成人,也有兒童病例的報道,其臨床表現與成人相似[3]。通常急性或亞急性起病,病變部位多位于腦、腦膜、脊髓及視神經,臨床表現主要為發熱、頭痛、腦病、不自主運動、癲癇、脊髓炎和視力異常等[1,2],其病因及發病機制不明。本文通過對宣武醫院2018年-2020年收治的5例GFAP星型膠質細胞病患者臨床資料進行分析,并復習相關文獻,探討GFAP患者的臨床特點,為臨床醫師早期識別和診斷提供參考。
1 對象與方法
1.1 研究對象 選取5例2018年-2020年經首都醫科大學宣武醫院診治的GFAP星型膠質細胞病患者,5例患者均行腰穿檢查,應用CBA檢測方法檢測腦脊液GFAP抗體陽性確診。
1.2 研究方法 回顧分析5例GFAP星型膠質細胞病患者的臨床表現、神經系統查體、腦脊液檢查、腦電圖、影像學檢查及治療方式。
2 結 果
2.1 臨床特點 收集的5例GFAP星型膠質細胞病患者中2例男性,3例女性。發病年齡最小為31歲,最大為78歲;病程多為亞急性起病,從50 d~6個月不等;臨床表現為肢體乏力(4例)、睡眠增多(3例)、發熱(3例)、記憶力下降(3例)、癲癇(1例)、肢體不自主抖動(1例)、肢體麻木(1例)、尿便障礙(4例)、視物模糊(1例),其中有1例以后頸枕及后背疼痛起病,1例出現視幻覺。
2.2 腦脊液結果 腦脊液常規檢查可見白細胞計數及蛋白含量不同程度的增高(見表1);腦脊液免疫學化驗檢查顯示多數患者出現腦脊液寡克隆區帶及腦脊液特異性寡克隆區帶陽性、24 h鞘內合成率不同程度的增高(見表2)。
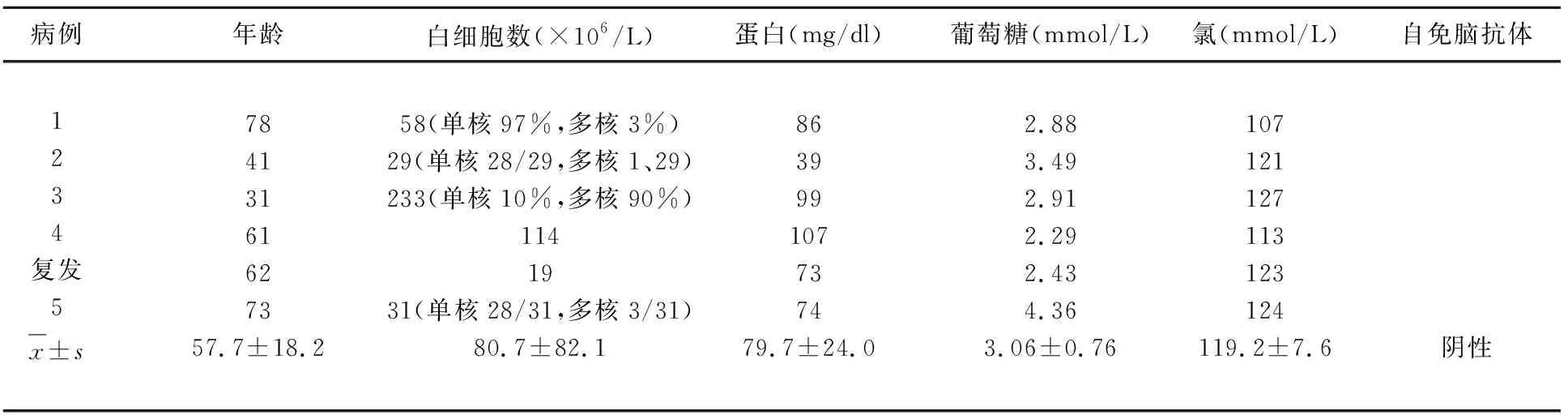
表1 腦脊液常規檢查結果
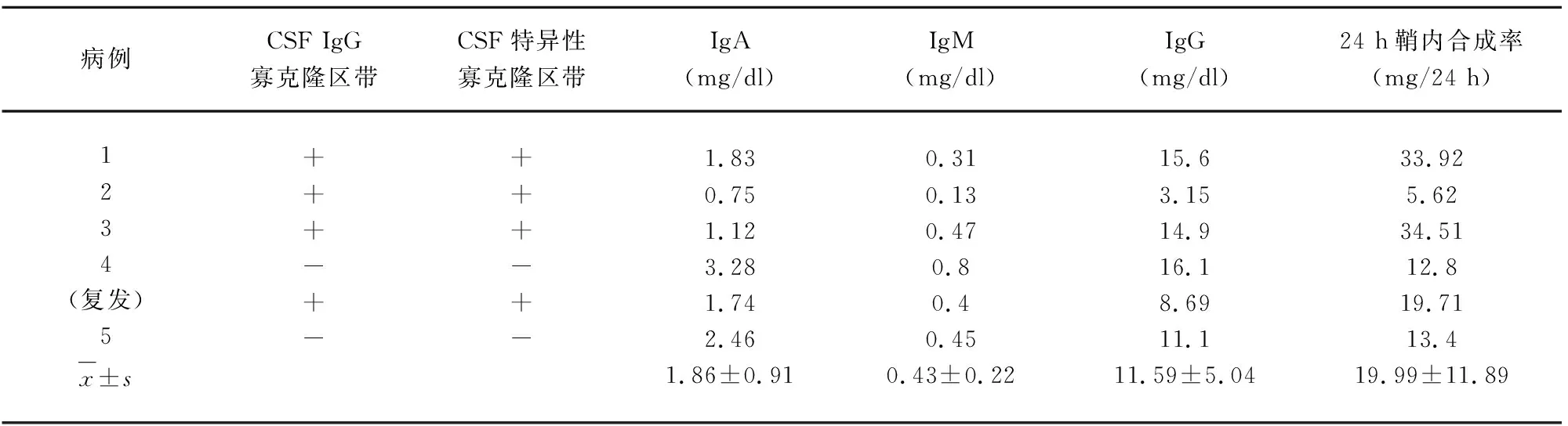
表2 腦脊液免疫學化驗結果
2.3 腦電圖結果 病例1:彌漫性異常視頻腦電圖(背景為慢活動,尖波、尖慢復合波、不典型三相波混雜);病例2:背景中混有稍多低幅慢波;病例3:未做;病例4:中度異常,全導可見大量中至高幅4~5 Hz慢波;病例5:全導較多低中幅慢波及快波,蝶骨偶見可疑尖波,符合腦炎表現。
2.4 影像學檢查結果 病例1:頭部核磁:雙側基底節區、側腦室旁、半卵圓中心多發腔隙性腦梗死,部分軟化灶,右側上頜竇炎,右側小腦半球、基底節區微出血灶,雙側海馬萎縮;胸腰椎核磁:胸腰髓未見明確病變。病例2:頭部核磁:左側顳葉、雙側側腦室后角旁及胼胝體異常信號影,考慮:非腫瘤性病變,不除外腦炎;頸、胸、腰椎核磁:頸5-胸2水平脊髓內異常信號影;腰髓未見異常信號;應用激素治療后復查:頭部核磁:雙側腦室周圍白質異常信號,左側為著。頸、胸椎核磁:脊髓內未見明確病變。病例3:頭部核磁:未見明顯異常;頸髓+胸髓+腰椎核磁:C5-7、T7、T8水平脊髓可疑異常信號,炎癥待排;腰髓未見明顯異常;激素治療后復查:頸椎核磁:延髓、頸6、頸7平面脊髓異常信號,缺血?胸髓核磁:胸5水平髓內異常信號。病例4:初次發病:頭部核磁:雙側內顳葉異常信號,左側腦室旁及左額葉皮質下缺血灶,輕度腦白質變性;頸髓核磁:延髓-胸1水平脊髓條片狀異常信號;停藥后復發所見:胸髓平掃+增強:延髓-胸10水平脊髓異常信號(見圖1);頭部核磁:雙側內顳葉、左側島葉、橋腦、雙側基底節、側腦室周圍異常信號(見圖2)。病例5:頭部核磁:左側海馬鉤回、前連合、雙額葉內側、扣帶回多發異常信號;頸胸核磁:頸髓及胸髓內未見異常信號。
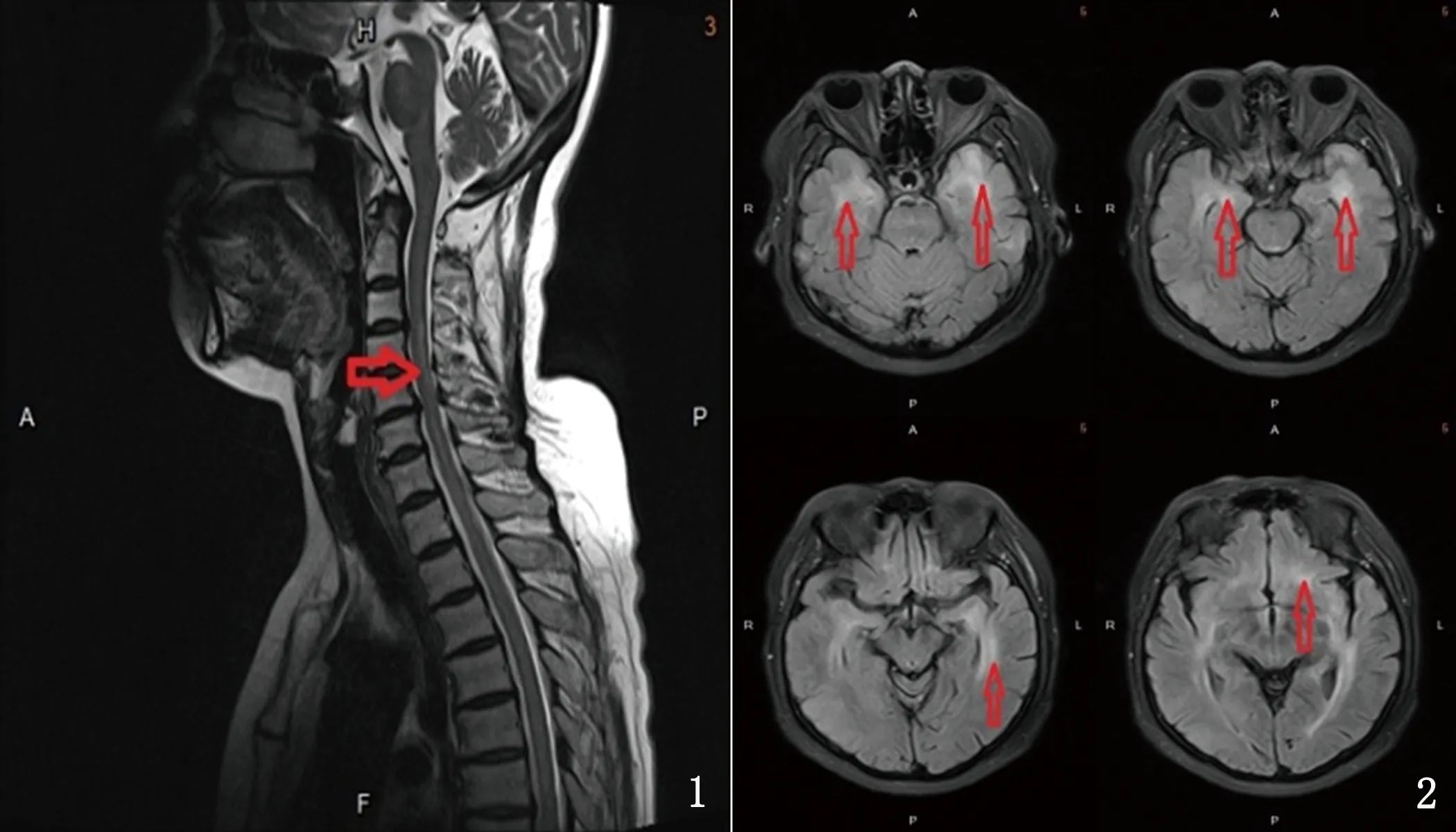
圖1 延髓-胸10水平脊髓異常信號;圖2:雙側內顳葉、左側島葉、橋腦、雙側基底節、側腦室周圍異常信號
2.5 治療及預后 5例患者均給予激素治療,癥狀均不同程度改善(見表3)。

表3 治療方案及預后
3 討 論
自身免疫性膠質纖維酸性蛋白(GFAP)星型細胞病是一種少見的炎性中樞神經系統疾病,目前關于本病的診斷尚無統一標準,主要診斷要點有:(1)急性或亞急性起病,臨床表現為腦膜、腦、脊髓、視神經受累或各種癥狀的組合;(2)典型MRI可見腦室旁線樣放射狀強化和(或)脊髓長節段受累伴中央強化;(3)腦脊液GFAP抗體陽性;(4)腦活體組織檢查提示小血管周圍炎癥伴小膠質細胞活化;(5)類固醇激素治療有效;(6)排除其他可能疾病;在我們搜集的這5例患者中,發病年齡從31~78歲不等,男性2例,女性3例,病程最短者50 d,最長者6個月。腦脊液檢查顯示白細胞計數及蛋白定量均升高、免疫學化驗異常,證實了GFAP星型膠質細胞病為免疫機制介導的炎性中樞神經系統病變。這與文獻報道的相一致[2]。我們搜集的這5例患者中,第2例和第5例患者腦脊液及血清中均檢測到GFAP-IgG抗體,第2例患者臨床表現為頸及后背疼痛、意識障礙、行走困難及雙手麻木,當地醫院考慮為頸椎病,未給予特殊治療,后逐漸出現雙手不自主抖動,就診于當地醫院,發現血糖增高,于內分泌科住院治療,血糖控制良好出院,但雙手不自主抖動未見明顯好轉。出院后患者逐漸出現意識模糊、不認識家人、語無倫次,間斷發熱,伴行走困難,再次于當地醫院住院治療,完善腰穿檢查顯示腦脊液蛋白及細胞數均升高,行頭部及脊髓MRI檢查示左側顳葉、雙側側腦室后角旁及胼胝體異常信號,不除外腦炎,頸5及胸2脊髓內可見長節段異常信號,考慮炎性脫髓鞘病變。當地醫院給予激素治療,患者癥狀明顯好轉。后患者因出現雙手麻木就診于我院,化驗腦脊液及血清GFAP-IgG抗體陽性,繼續予以激素治療,患者癥狀好轉出院。第5例患者臨床表現為乏力、睡眠增多及反應遲鈍,首先于內分泌科就診,血糖控制良好出院。后患者癥狀仍進行性加重,就診于我院,完善頭部核磁示:海馬鉤回、前連合、雙額葉內側、扣帶回多發異常信號,腦脊液及血清GFAP-IgG抗體陽性,給予患者激素沖擊治療,癥狀好轉出院。以上2例患者的診治過程提示GFAP星型膠質細胞病患者的臨床癥狀缺乏特異性,患者可因肢體乏力就診于多個科室,從而延誤了患者的正規診治,也增加了患者的經濟負擔。另外這2例患者均存在血糖異常,而其余3例腦脊液GFAP-IgG抗體陽性,血清陰性的患者,血糖均正常,提示血清GFAP抗體可作為預測自身免疫性糖尿病的新型生物學標志物,這與文獻報道的一致[4]。
GFAP-IgG抗體有時與NMDA-R IgG、AQP4-IgG及MOG共存,此種情況常見于合并畸胎瘤的患者。Lennon等人的研究顯示,34%的患者合并腫瘤,這其中66%的患者在神經系統發病2 y內出現[1,2]。最常見的腫瘤為卵巢畸胎瘤,其他少見腫瘤包括腺癌(乳腺、肺、卵巢、子宮內膜、食道癌和腎癌)、頭頸部鱗狀細胞癌、胸腺瘤、多形性腮腺瘤、膠質瘤、多發性骨髓瘤、小細胞癌和類癌[2,5~7]。我們搜集的這5例患者,全部送檢NMDA-R、AQP4及MOG相關抗體檢測,結果均為陰性,且到目前為止未發現腫瘤的存在,也可能與病例數少,隨訪時間尚短有關。其它非神經元抗體包括抗核抗體(ANA)、抗干燥綜合征抗體(SSA及SSB)及抗雙鏈DNA抗體等[5]。本組病例1化驗抗核抗體譜示:Nrnp/Sm抗體陽性()、抗Sm抗體弱陽(+)、抗Ro-52抗體強陽()。病例5抗核抗體結果為核顆粒型1∶100。提示免疫因素在GFAP星型細胞病中起了一定作用,但具體機制目前尚不清楚,有待進一步研究。
GFAP星型細胞病患者的影像學表現多為非特異性的,最為常見的是沿中線分布的T2加權像上高信號的病灶,增強后可見病灶有強化,其次為軟腦膜、室管膜強化。特征性的影像學改變為垂直于腦室的血管周圍線性放射狀強化,脊髓病變多為長節段脊髓炎[1,6]。且這種強化在經過治療后會消失,病理顯示為腦膜炎和小血管周圍炎,提示強化是由于釓從受損的血腦屏障滲漏所致[5]。治療后,血腦屏障迅速修復,強化消失。腦PET顯像可顯示與MRI異常區域相對應的高代謝。我們報道的第2例患者,在應用激素治療后復查脊髓核磁病灶消失,復查頭部核磁病灶較前明顯縮小,第5例患者行PET檢查顯示:左側海馬溝回、前聯合、雙側額葉內側、扣帶回異常信號葡萄糖代謝增高,考慮顱內原發病變(炎性?)。與文獻報道一致。與AQP4-IgG病變相比,GFAP星型膠質細胞病患者的脊髓病變病變邊界比較模糊,脊髓腫脹較少見,且強化主要表現為脊髓中央管斑點狀強化或軟腦膜強化。
腦電圖表現缺乏特異性,本組搜集的5例患者中有4例腦電圖均表現為不同程度的慢波,這4例患者頭部核磁均證實有顱內病變,提示腦電圖異常與顱內受累部位之間可能有一定關聯性。
GFAP患者的治療目前尚無統一的標準或共識。急性期治療一般包括大劑量糖皮質激素沖擊、靜脈注射免疫球蛋白和血漿置換等。長期治療包括口服類固醇和免疫抑制劑。大約70%的患者對類固醇治療反應良好。部分患者在激素減量過程中或停藥后復發。本組搜集的5例患者起始均應用了大劑量甲強龍沖擊治療,后逐漸序貫為口服治療,出院時癥狀均有不同程度的好轉,病例3以小便困難及雙下肢無力入院,病程中有癲癇發作,經激素沖擊治療后,患者未再出現癲癇發作,但該患者雙下肢無力癥狀未見改善,且該患者肌電圖檢查示周圍神經損害,這在既往文獻中未見明確報道,是否與GFAP抗體有關,有待于進一步探討。另外,病例4第一次發病后在潑尼松減量至10 mg時自行停藥致疾病復發,再次入院后重新啟動大劑量甲強龍沖擊治療,癥狀再次好轉,提示本病對大劑量糖皮質激素較敏感,病情平穩后仍不宜快速停藥。但藥物應用的總療程目前尚無統一定論。
綜上所述,GFAP星形細胞病是一種神經系統自身免疫性疾病,其病因、病理、機制、診斷和治療等方面有待進一步研究。
猜你喜歡
鴨綠江(2021年35期)2021-04-19 12:24:18
考試與評價·高一版(2020年6期)2020-11-02 02:45:24
媽媽寶寶(2019年10期)2019-10-26 02:45:34
中國生殖健康(2019年3期)2019-02-01 06:12:26
鐵道通信信號(2018年11期)2019-01-19 01:15:08
電子制作(2018年11期)2018-08-04 03:25:42
鐵道通信信號(2018年2期)2018-04-18 12:18:10
鐵道通信信號(2016年11期)2016-06-01 12:11:32
鑿巖機械氣動工具(2016年3期)2016-03-01 04:00:25
中國病理生理雜志(2015年8期)2015-12-21 12:38:06
