磁性固相萃取結合GC-MS檢測煙葉中擬除蟲菊酯類農藥殘留
2021-08-06 09:37:42楊玉明劉妧晨劉智敏司曉喜劉宏程許志剛
分析測試學報 2021年7期
關鍵詞:復合材料
楊玉明,劉妧晨,劉智敏,李 嬌,司曉喜,林 濤,劉宏程,許志剛*
(1.昆明理工大學 理學院,云南 昆明 650500;2.云南中煙工業有限責任公司技術中心,云南 昆明 650231;
3.云南省農業科學院 質量標準與檢測技術研究所,云南 昆明 650223)
擬除蟲菊酯是20世紀70年代人工合成的一類殺蟲劑,因具有廉價、殺蟲譜廣、低毒性、低殘留、對環境友好等特點[1],被廣泛用于蔬菜、茶葉等農產品生產[2-3]。目前,擬除蟲菊酯類殺蟲劑已發展成為繼有機磷類農藥、氨基甲酸酯類農藥后使用最廣的第三大類殺蟲劑[4-5]。然而,已有研究發現擬除蟲菊酯類殺蟲劑在自然環境下降解速率慢,對光與熱具有一定的穩定性,環境殘留的擬除蟲菊酯類殺蟲劑會引發毒副作用[6-7]。此外,環境中殘留的擬除蟲菊酯類殺蟲劑會通過食物鏈富集進入機體,危害人等哺乳動物的心血管、免疫力和生殖系統等[8-9]。因此,擬除蟲菊酯類殺蟲劑的殘留問題受到普遍重視,如歐盟、美國等限定煙草中菊酯類農藥最高殘留量為0.5 mg/kg[10];我國國家標準YC/T 405.2-2011也將擬除蟲菊酯類殺蟲劑殘留量設定為煙草中的檢測項目并嚴格規范[11],但仍有煙草中檢出擬除蟲菊酯類農藥的報道[12-14]。
目前,擬除蟲菊酯類殺蟲劑殘留的檢測方法有高效液相色譜法[15]、毛細管氣相色譜法[16]、液相色譜-串聯質譜法[17]、氣相色譜-串聯質譜法[18]等。但煙草等農產品中存在擬除蟲菊酯類殺蟲劑含量低、基質復雜、分析物種類多以及化合物間結構與性質差異小等問題,分析前一般需進行固相萃取[19]、分散固相萃取[20]、分散液相微萃取[21]、磁固相萃取[22]等樣品前處理過程。其中磁性固相萃取(Magetic solid phase extraction,MSPE)是由?afa?íková等[23]于1999年在固相萃取技術的基礎上發展的一種高效樣品前處理方法,因吸附劑具有磁性,可在外加磁場中實現目標分析物與基質的快速分離,前處理操作簡單,且磁性吸附劑易于回收和重復利用。環糊精是一類天然環狀低聚糖化合物,具有疏水內腔和親水外殼,且外殼含有活性羥基,使得環糊精可與有機分子、無機分子等分析物發生吸附作用[24-25],已廣泛用于化工、食品、醫療等領域。環糊精作為吸附劑還需一定基質,使其可重復利用與回收[26]。
本研究選擇無機材料SiO2為吸附劑基質,采用水熱反應法制備了磁性Fe3O4納米粒子,涂覆SiO2后,通過烷基偶聯試劑γ-縮水甘油醚氧丙基三甲氧基硅烷鍵合上β-環糊精,得到Fe3O4@SiO2@β-CD磁性固相萃取劑,結合氣相色譜-質譜實現了煙草樣品中4種擬除蟲菊酯類殺蟲劑殘留的分析。
1 實驗部分
1.1 儀器與試劑
Clarus 600 GC氣相色譜儀、Clarus 600 MS質譜檢測器(美國珀金埃爾默公司);FV-SK-02型渦旋振蕩器(美國萊伯特公司);KQ-700V型超聲波清洗機(昆山市超聲儀器公司);水熱反應釜(含聚四氟乙烯內膽,南京瑞尼克科技有限公司);NDO-400型定溫恒溫干燥箱(上海愛朗儀器有限公司);79HW-1電子調溫模擬控溫磁力加熱攪拌器(江蘇優卓諾儀器制造有限公司);BT125D型電子天平儀器(德國賽多利斯科學儀器有限公司);Nova NanoSem 450掃描電鏡(美國FEI公司);TENSOR27紅外光譜儀(德國Bruker公司);PANayltical Empyrean型X射線衍射儀(PANalytical公司);AUTOSORBIQ-MP微孔吸附材料分析測試儀(美國康塔儀器公司);0.22μm尼龍有機系濾膜(上海新亞凈化器件廠)。
FeCl3·6H2O(98%)、乙二醇(AR,98%)、醋酸鈉(AR,99%)、聚乙二醇(PEG-2000)(Average Mn2000)、γ-縮水甘油醚氧丙基三甲氧基硅烷(KH560,98%)均購于阿拉丁公司;甲醇、乙腈等溶劑購于四川西隴化工有限公司;色譜級甲醇和乙腈等溶劑購于天津市津東天正精細化學試劑廠;NH3·H2O(25%)和N,N-二甲基酰胺(DMF)購于天津市風船化學試劑科技有限公司;工業氮氣購于昆明融泰工貿有限公司;純凈水購于杭州娃哈哈集團有限公司;丙酮購于重慶川東化工(集團)有限公司;鹽酸(36%~38%)購于云南楊林工業開發區汕滇藥業有限公司;乙醇(分析純)購自天津市致遠化學試劑有限公司;氫化鈉(NaH,80%)購自山東西亞化學股份有限公司;α-CD、β-CD、γ-CD購于山東濱州智源生物有限公司;聯苯菊酯、甲氰菊酯、氟氯氰菊酯、溴氰菊酯4種標準品純度大于95%均購于阿拉丁公司;煙草葉片采自云南紅河州當地。
1.2 環糊精磁性復合材料的制備
準確稱取2.7 g FeCl3·6H2O溶于50 mL乙二醇(EG)中,室溫劇烈攪拌30 min后,再加入7.2 g醋酸鈉和2.0 g聚乙二醇(PEG-2000),繼續攪拌1 h,然后將溶液轉移至含有聚四氟乙烯襯里的不銹鋼高壓釜中,密封后于200℃下加熱8 h,將溶液中生成的黑色沉淀物冷卻至室溫并依次用乙醇和超純水洗滌數次直至溶液pH值呈中性,得到的Fe3O4粉末于60℃下真空干燥。
準確稱取1.0 g Fe3O4,加入10 mL 0.1 mol/L鹽酸,超聲處理10 min,通過磁分離除去液體,待固體物質上殘留液體自然揮干,向有黑色顆粒的燒杯添加80 mL乙醇,再加入20 mL超純水和2.5 mL NH3·H2O,攪拌均勻后,快速加入0.5 mL四乙氧基硅烷(TEOS)并在室溫下攪拌12 h,得到Fe3O4@SiO2。
精確稱取1.5 gβ-環糊精(使用前于90℃真空干燥8 h)溶于50 mL無水N,N-二甲基甲酰胺(DMF)中,加入0.15 g氫化鈉并于室溫下攪拌30 min,濾去未反應的固體物質,向濾液中加入0.5 gγ-縮水甘油醚氧丙基三甲氧基硅烷(KH560),氮氣保護下于90℃反應5 h,再加入2.0 g Fe3O4@SiO2,提高溫度至110~120℃繼續反應24 h,將反應液冷卻至室溫,過濾,產物依次用DMF、二次蒸餾水、甲醇和丙酮洗滌4~5次,濾干后,將產物于100℃真空干燥24 h,得黑色固體Fe3O4@SiO2@β-CD,制備流程見圖1。相同條件下制備Fe3O4@SiO2@α-CD和Fe3O4@SiO2@γ-CD用于萃取對照試驗。
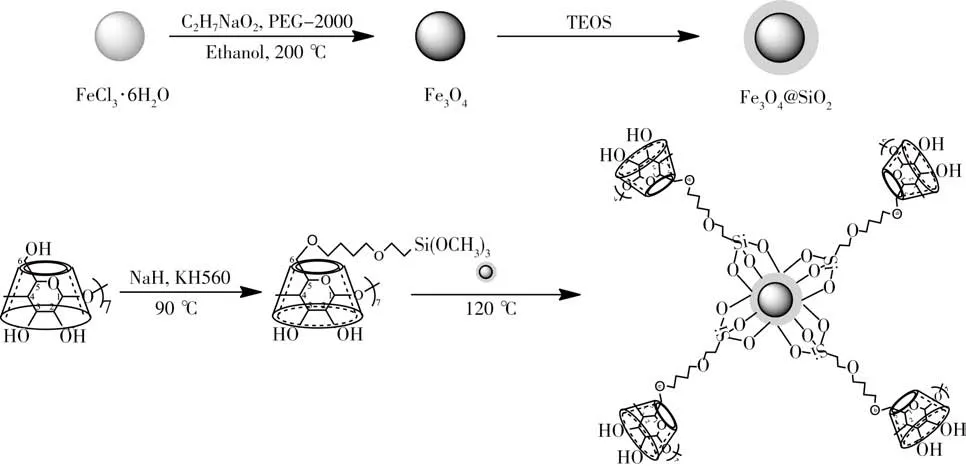
圖1 β-環糊精磁性復合材料的制備流程圖Fig.1 Diagram of preparation ofβ-cyclodextrin magnetic composite materials
1.3 樣品前處理
煙草煙葉樣品由云南中煙工業有限責任公司技術中心提供,3種煙葉樣品經初烤后參考文獻處理[27]:先將樣品粉碎,準確稱取100 mg于50 mL離心管中,加入2 mL甲醇,超聲處理30 min,將目標分析物從煙草煙葉組織中提取到甲醇溶液中,超純水定容至50 mL,并加入50 mg Fe3O4@SiO2@β-CD吸附劑,以150 r/min振蕩提取120 min。磁分離棄上層溶液,待吸附材料中含有的少量液體自然揮干后加入2 mL甲醇(色譜純),超聲解吸5 min,再次磁分離得上層清液,過0.22μm尼龍有機相濾膜后,待GC-MS測定,每種樣品平行測定3次。
1.4 色譜-質譜條件
DB-5 MS彈性石英毛細管色譜柱(30 m×0.25 mm i.d.×0.25μm d.f.,美國Agilent公司);進樣口溫度300℃,載氣為氦氣(純度≥99.999%),恒流流速1.0 mL/min;進樣量1μL,分流進樣,分流比10∶1。程序升溫:初始溫度150℃,保持1 min,以10℃/min升至210℃,再以3℃/min升至300℃,保存5 min。電離方式:電子源轟擊(EI);電子能量:70 eV;離子源溫度:200℃;傳輸線溫度:280℃;溶劑延遲:5.50 min;選擇離子模式(SIM)檢測。
2 結果與討論
2.1 環糊精磁性復合材料的結構表征
2.1.1 電鏡表征 采用掃描電鏡對制備的Fe3O4、Fe3O4@SiO2和Fe3O4@SiO2@β-CD進行表征。結果顯示:Fe3O4鍵合SiO2和β-CD后,表面結構和小球尺寸均發生了明顯變化。結果顯示:裸露的Fe3O4表面比較均勻且呈球形,平均直徑約214.8 nm(圖2A)。經SiO2修飾后,尺寸增大至227.6 nm得到Fe3O4@SiO2(圖2B),再經β-CD修飾后尺寸為263.3 nm(圖2C),修飾后的小球表面明顯比Fe3O4更粗糙、多孔,由此表明SiO2和β-CD已鍵合在Fe3O4表面成功制備了Fe3O4@SiO2@β-CD復合材料。

圖2 Fe3O4(A)、Fe3O4@SiO2(B)和Fe3O4@SiO2@β-CD(C)的掃描電鏡圖Fig.2 Scanning electron microscope pictures of Fe3O4(A),Fe3O4@SiO2(B)and Fe3O4@Si O2@β-CD(C)
2.1.2 紅外光譜表征β-CD、Fe3O4、Fe3O4@SiO2和Fe3O4@SiO2@β-CD的紅外光譜圖見圖3。由β-CD紅外光譜可見,3 343 cm-1處為環糊精表面羥基的伸縮振動峰,2 921 cm-1處為環糊精CH2伸縮振動吸收峰。Fe3O4紅外光譜中586 cm-1和3 300~3 500 cm-1處的寬峰分別為Fe―O的特征吸收峰及其表面羥基的伸縮振動峰。Fe3O4@SiO2和Fe3O4@SiO2@β-CD存在1 105 cm-1和1 111 cm-1處拉伸的特征硅峰Si―O―Si,表明二氧化硅外殼已形成。此外,Fe3O4@SiO2和Fe3O4@SiO2@β-CD的其他特征吸收帶Si―OH拉伸振動吸收峰、Si―O彎曲振動吸收峰、Si―O―Si彎曲振動吸收峰分別為960、795、475 cm-1和958、794、474 cm-1,Fe―O的特征峰從586 cm-1移至582 cm-1,表明二氧化硅殼通過Fe―O―Si化學鍵連接到磁性Fe3O4納米顆粒表面。Fe3O4@SiO2@β-CD的光譜圖中,2 850 cm-1和2 924 cm-1處存在信號增強的不對稱和對稱的C―H伸縮振動吸收峰,且峰的重疊導致信號增強,表明β-CD與Fe3O4@SiO2已成功接枝形成Fe3O4@SiO2@β-CD材料。
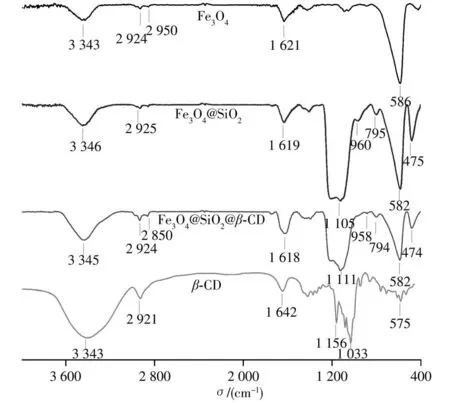
圖3 β-CD、Fe3O4、Fe3O4@SiO2和Fe3O4@SiO2@β-CD的紅外光譜圖Fig.3 FT-IR spectra ofβ-CD,Fe3O4,Fe3O4@Si O2 and Fe3O4@SiO2@β-CD
2.1.3 X射線衍射表征β-CD、Fe3O4、Fe3O4@SiO2和Fe3O4@SiO2@β-CD吸附材料進一步采用X射線衍射(XRD)表征(圖4)。結果顯示:磁性Fe3O4在29.2°、35.6°、43.4°、53.5°、57.1°和62.7°出現特征衍射峰,分別對應Fe3O4的(220)、(311)、(400)、(422)、(511)、(440)晶面的衍射峰(JCPDS No 19-0629),當涂覆SiO2后,在16°~26°處出現非晶體SiO2導致的微弱寬衍射峰,無尖銳的衍射峰,這是因為涂覆過程采用的是TEOS,因而在Fe3O4表面生成的是無定形硅基材料,所以SiO2衍射峰不明顯,僅在18.14°出現對應的非晶體態硅殼的弱衍射峰[28]。進一步在Fe3O4@SiO2表面鍵合環糊精,于6.1°處出現β-CD的有關特征峰,表明環糊精已成功鍵合在Fe3O4@SiO2表面。
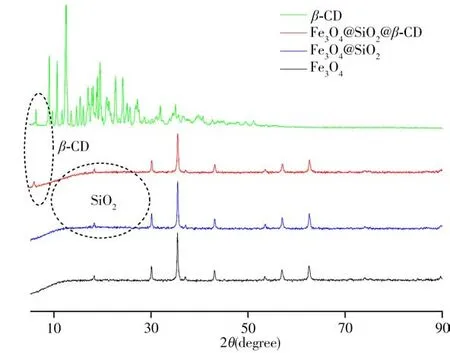
圖4 β-CD、Fe3O4、Fe3O4@SiO2和Fe3O4@SiO2@β-CD的XRD光譜圖Fig.4 XRD spectra ofβ-CD,Fe3O4,Fe3O4@SiO2 and Fe3O4@SiO2@β-CD
2.1.4 N2吸附-脫附表征 由Fe3O4、Fe3O4@SiO2和Fe3O4@SiO2@β-CD復合材料樣品在77 K下的N2吸附等溫線和孔徑分布圖可見(圖5A),三者均含有明顯的滯后環,這是典型的Ⅳ型等溫線,當相對壓力(P/Po)小于0.1時,材料對氮氣的吸附量急劇增加,表明在較低壓力下,復合材料和氮氣的相互作用力大,Fe3O4@SiO2@β-CD等復合材料含有微孔,而回滯環的存在可能是由于Fe3O4、Fe3O4@SiO2和Fe3O4@SiO2@β-CD納米材料尺寸太小(≈200 nm)且具有磁性作用,使得納米粒子間相互吸附產生中孔和大孔的形狀結構,從而與氮氣發生相互作用所致。從Fe3O4、Fe3O4@SiO2和Fe3O4@SiO2@β-CD復合材料的孔徑分布圖和不同孔徑的分布情況可見(圖5B、C),在Fe3O4上逐步涂敷SiO2和β-CD,合成的Fe3O4@SiO2和Fe3O4@SiO2@β-CD孔隙尺寸變大,3種材料的大孔比例分別為49.42%、49.85%和98.17%,其比表面積分別為25.044、21.046、15.374 m2/g,孔體積分別為0.134、0.745、0.993 mL/g,平均孔徑分別為1.143、2.141、2.582 nm,由此可見,Fe3O4逐步涂敷SiO2和β-CD后,平均孔徑進一步增加。因此,更大的孔可能使Fe3O4@SiO2@β-CD相對于Fe3O4@SiO2和Fe3O4能更好地吸附目標分析物。
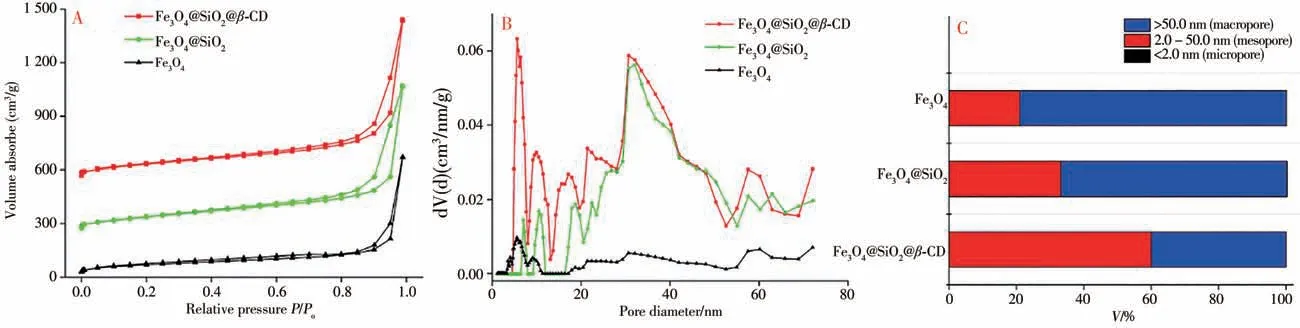
圖5 Fe3O4、Fe3O4@Si O2和Fe3O4@Si O2@β-CD的氮氣吸附-脫附等溫線和孔徑分布圖Fig.5 Nitrogen adsorption-desorption isotherm and pore size distribution map of Fe3O4,Fe3O4@SiO2 and Fe3O4@SiO2@β-CD
2.2 萃取條件的優化
為進一步提高吸附劑的萃取效率,實驗優化了萃取時間、解吸時間、解吸溶劑、萃取液pH值和鹽離子濃度等影響因素。將50 mg Fe3O4@Si O2@β-CD加入50 mL含有100μg/L聯苯菊酯、甲氰菊酯、氟氯氰菊酯和溴氰菊酯的混合水溶液中,通過回收率及解吸率參數考察材料的吸附萃取性能,具體公式為:E=m1/(m1+m2+m3+m4+…)×100%;R=me/m0×100%;式中E為解吸率,m1為第一次解吸后目標物的質量(μg),m2是第二次解吸后目標物的質量(μg),依次解吸直至無目標分析物檢出;R為萃取率,me為吸附后目標物的質量(μg),m0為吸附前溶液中目標物的質量(μg)。
2.2.1 萃取吸附劑的選擇 分別采用制備的Fe3O4@SiO2@α-CD、Fe3O4@SiO2@β-CD和Fe3O4@SiO2@γ-CD萃取100μg/L的聯苯菊酯、甲氰菊酯、氟氯氰菊酯和溴氰菊酯混合水溶液,考察了3種環糊精衍生物(α-CD、β-CD和γ-CD)復合材料吸附性能的差異。結果顯示:3種環糊精磁性材料均能吸附4種擬除蟲菊酯類化合物,但鍵合β-CD的吸附劑對所有分析物的萃取率最大,因此選擇Fe3O4@SiO2@β-CD為磁性固相萃取吸附劑。
2.2.2 萃取實驗參數的優化 實驗考察了萃取時間、解吸時間、解吸溶劑、萃取溶液pH值及離子強度對Fe3O4@SiO2@β-CD吸附4種擬除蟲菊酯類物質的影響。結果顯示,改性后的磁性材料可在30 min內對4種物質達到吸附平衡,因此選擇最佳萃取時間為30 min。進一步研究了Fe3O4@SiO2@β-CD材料在不同時間(2~20 min)超聲解吸對4種擬除蟲菊酯類物質萃取效率的影響,發現均可在15 min達到解吸平衡。另外,由于擬除蟲菊酯類物質在不同解吸溶劑中有不同的分配系數,因此實驗考察了甲醇、乙腈、乙醇、異丙醇、二氯甲烷5種不同解吸溶劑對萃取效率的影響。結果顯示:當解吸溶劑為甲醇時,聯苯菊酯、甲氰菊酯、氟氯氰菊酯和溴氰菊酯有最大的萃取效率,因此選擇甲醇為最優解吸溶劑。由于目標分析物在不同pH值溶液中存在的形態不同,從而影響分析物的萃取效率,因此實驗考察了不同pH值(2.0、4.0、6.0、7.0、8.0、10.0、12.0)對萃取效率的影響,發現在酸性或堿性條件下,萃取效率明顯下降,這可能是由于擬除蟲菊酯類物質在過酸過堿的水溶液中解離,從而導致萃取效率下降。因此,萃取溶液的最佳pH值選為7.0。在提取過程中加入適量NaCl也會影響提取效率,且4種擬除蟲菊酯類物質的萃取效率隨離子強度的增加而下降,這是因為磁性復合材料中β-CD的空腔對Na+也有一定包結作用,使得Fe3O4@SiO2@β-CD與4種擬除蟲菊酯類物質的相互作用力減弱,導致吸附效率降低。因此,萃取液中不添加NaCl更有利于材料對目標物的吸附分離。
2.2.3 萃取容量 實驗通過萃取容量考察了Fe3O4、Fe3O4@SiO2和Fe3O4@SiO2@β-CD的吸附能力,在最佳萃取條件下,萃取質量濃度為100~4 000μg/L的擬除蟲菊酯混合溶液,其萃取量為Q/(C0-CR)×V/M,式中Q為分析物的萃取量,C0為解吸后得到的質量濃度(μg/L),CR為吸附前溶液中目標物質量濃度(μg/L),V為解吸后的進樣體積,M為Fe3O4@SiO2@β-CD復合材料的質量(g)。結果顯示,未經修飾的裸露四氧化三鐵由于表面相對光滑,與擬除蟲菊酯類物質作用的活性位點相對較少,因此吸附量較低;而在Fe3O4表面涂敷二氧化硅材料后使得材料表面增加了可吸附擬除蟲菊酯類物質作用的活性位點,因此Fe3O4@SiO2對4種擬除蟲菊酯類物質的吸附量有所增加;繼續鍵合β-CD后形成的Fe3O4@SiO2@β-CD的環糊精的空腔結構與擬除蟲菊酯類化合物的分子大小接近,進一步增加了對擬除蟲菊酯類物質的作用位點,因此Fe3O4@SiO2@β-CD復合材料對擬除蟲菊酯類物質的吸附量遠大于未經修飾的Fe3O4和Fe3O4@SiO2。
2.3 分析方法的建立
以Fe3O4@SiO2@β-CD為磁固相萃取吸附材料,在優化條件下采用GC-MS分析了聯苯菊酯、甲氰菊酯、氟氯氰菊酯和溴氰菊酯4種擬除蟲菊酯類殺蟲劑。結果顯示:在0.1~3.0 mg/kg含量范圍內,4種擬除蟲菊酯類殺蟲劑的響應信號與對應濃度呈良好的線性關系,相關系數(r2)為0.998 1~0.999 8,以3倍信噪比(S/N≥3)計算得其檢出限(LOD)均為0.03 mg/kg,以S/N≥10計算得定量下限(LOQ)均為0.1 mg/kg(表1),完全滿足煙葉中4種擬除蟲菊酯檢測靈敏度要求,總離子流圖見圖6。
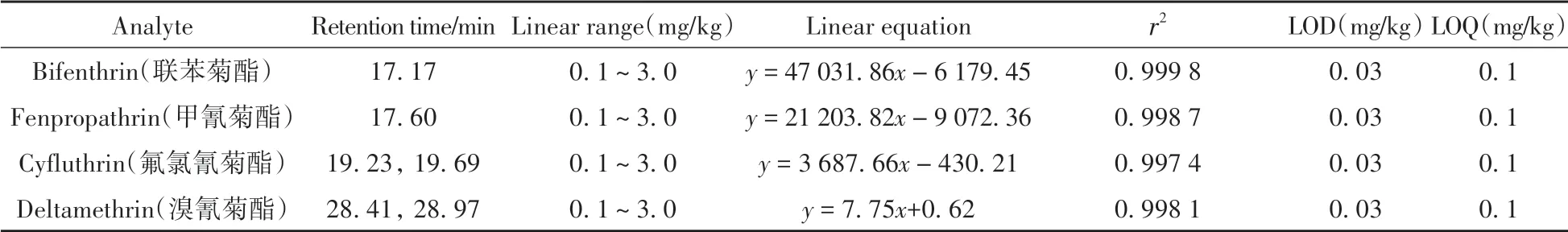
表1 4種擬除蟲菊酯類農藥的線性方程、相關系數(r2)、檢出限和定量下限Table 1 Linear equations,correlation coefficients(r2),LODs and LOQs of four pyrethroid pesticides
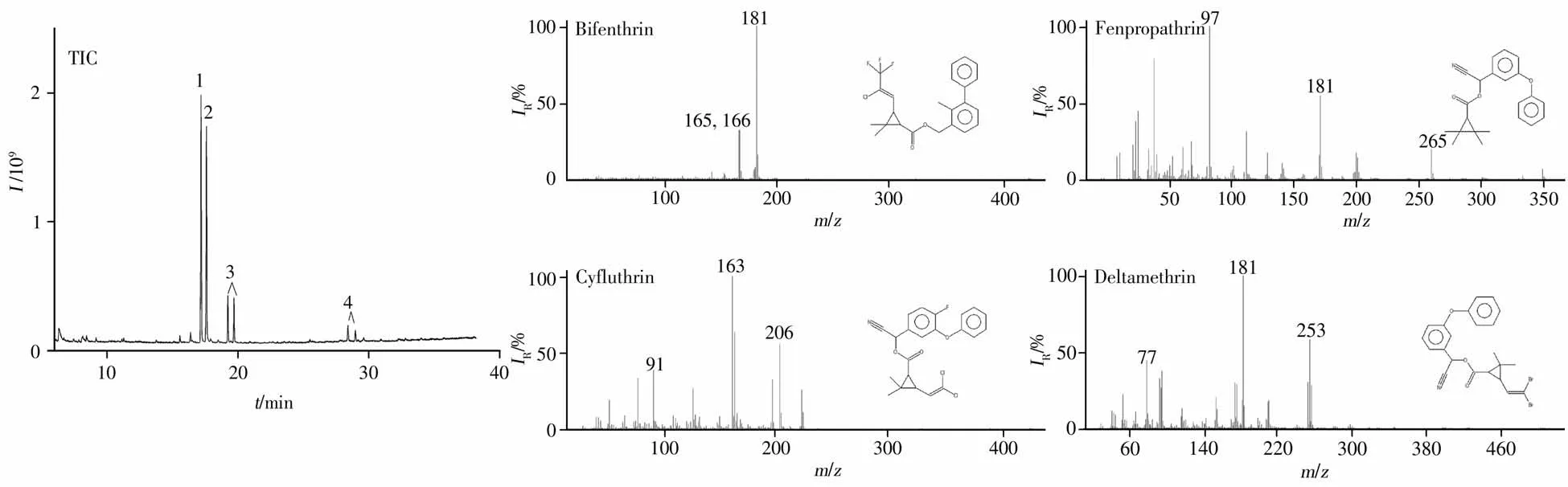
圖6 4種擬除蟲菊酯總離子流圖及其質譜圖Fig.6 Total ion chromatogram and mass spectra of four pyrethroids 1:bifenthrin,2:fenpropathrin,3:cyfluthrin,4:deltamethrin
2.4 實際樣品分析及加標回收率
采用本方法在優化條件下對10批市售煙葉樣品進行分析,結果在5批煙葉樣品中檢出聯苯菊酯、氟氯氰菊酯和溴氰菊酯農藥殘留,含量分別為1.08~1.78、0.10~0.35、0.12~0.75 mg/kg,但均未檢出甲氰菊酯。根據2020年國際煙草科學研究合作中心(CORESTA)農藥限量名單和2006年農用化學品咨詢委員會(ACAC)提出的現行法規中煙草中農藥的最大殘留限量標準(聯苯菊酯、氟氯氰菊酯和溴氰菊酯限量分別為3.00、2.00、1.00 mg/kg)[29],檢出的聯苯菊酯、氟氯氰菊酯和溴氰菊酯均未超標,其中一個陽性煙葉樣品色譜圖見圖7。
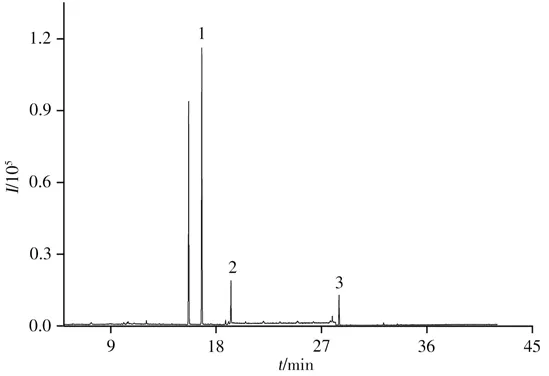
圖7 陽性煙葉樣品的選擇離子流圖Fig.7 Selected ion flow diagram of a positive tobacco sample 1:bifenthrin,2:cyfluthrin,3:deltamethrin
取另外5批陰性煙葉樣品(烤煙、白肋煙、香料煙、曬紅曬煙和曬黃曬煙)分別添加一定量4種擬除蟲菊酯類殺蟲劑,在優化條件下測定,計算方法的回收率及相對標準偏差(RSD)。結果顯示:4種擬除蟲菊酯類殺蟲劑的回收率為65.9%~107%,RSD為0.70%~12%(表2)。表明方法具有較好的準確性和精密度,可用于實際煙葉樣品中聯苯菊酯、甲氰菊酯、氟氯氰菊酯和溴氰菊酯4種擬除蟲菊酯類殺蟲劑的檢測。
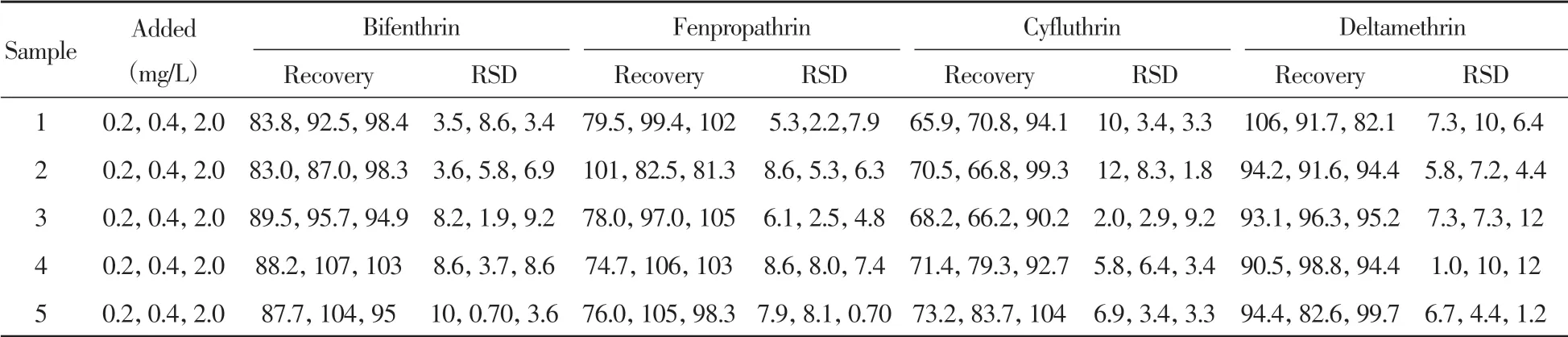
表2 不同煙草中4種擬除蟲菊酯類殺蟲劑的回收率的測定(n=3)Table 2 Determination of recovery of four pyrethroid pesticides in different tobaccos(n=3) (%)
3 結 論
本文將環糊精結合磁性納米Fe3O4顆粒得到具有磁性的環糊精磁性復合材料,并聯用氣相色譜-質譜建立了煙葉中聯苯菊酯、甲氰菊酯、氟氯氰菊酯和溴氰菊酯4種擬除蟲菊酯類殺蟲劑的快速分析方法,方法的檢出限為0.03 mg/kg,定量下限為0.1 mg/kg,回收率為65.9%~107%,RSD為0.70%~12%。該環糊精磁性復合材料具有制備簡單,樣品前處理易于操作,萃取效率高且易分離等優點,可用于煙葉樣品中擬除蟲菊酯類殺蟲劑農藥殘留的檢測分析。
猜你喜歡
建材發展導向(2022年2期)2022-03-08 01:44:04
建材發展導向(2021年14期)2021-08-23 00:56:16
中國材料進展(2019年10期)2019-12-07 05:32:14
纖維復合材料(2018年3期)2018-04-25 07:22:58
電子測試(2017年11期)2017-12-15 08:57:13
山東工業技術(2016年15期)2016-12-01 05:31:34
中國塑料(2015年6期)2015-11-13 03:02:54
中國塑料(2015年11期)2015-10-14 01:14:14
中國塑料(2015年8期)2015-10-14 01:10:41
應用化工(2014年10期)2014-08-16 13:11:29
