雙配體CuFe@MOFs材料為前驅體的催化劑的組分調控對CO2加氫制C2+醇性能的影響
2021-08-10 08:33:52溫月麗范鶴鳴宋镕鵬張維中
無機化學學報 2021年8期
張 倩 溫月麗*, 王 斌 范鶴鳴 楊 晨 宋镕鵬 張維中 黃 偉*,,3
(1太原理工大學環境科學與工程學院,太原 030024)
(2省部共建煤基能源清潔高效利用國家重點實驗室,太原 030024)
(3山西太原理工煤轉化技術工程有限公司,太原 030024)
0 引言
人類對化石燃料的過度依賴不僅導致能源短缺,還引發了因CO2過度排放所帶來的一系列環境問題[1-2]。CO2減量化、資源化利用已引起國際社會的廣泛關注[3-4]。習近平總書記在2020年召開的聯合國大會上也倡導通過“人工碳匯”實現“碳中和”目標。
CO2催化加氫轉化為燃料或其它高附加值化學品,是固定化、資源化利用CO2的經濟有效的途徑,可以實現“資源—CO2—資源”的可持續循環利用,具有重要的戰略意義[5-7]。相比于甲醇,加氫產物中的C2+醇合成需要同時控制CO2還原和碳鏈的增長[8-9],更具有挑戰性。
當前制約CO2加氫合成C2+醇工業化的因素主要是催化劑催化效率低、壽命短、穩定性差等問題。在催化劑的研究中,改性費托催化劑[10-11]因原料易得、價格低廉頗受關注。其中,相比于穩定性差、醇選擇性低的CuCo催化劑,CuFe雙組分催化劑更具催化優勢,因為其中的Fe可以同時催化逆水煤氣變換(RWGS)和費托合成(FTS)反應[12],Cu 既能活化 H2又對CO非解離插入具有活性。然而,銅活性組分在高溫加氫條件下容易發生遷移,引起顆粒團聚、燒結[7],導致催化劑活性和穩定性差[13],在一定程度上阻礙了CuFe基催化劑的應用。目前的研究均集中于引入載體或助劑以期提高金屬的分散性、相互作用、酸堿性進而提高其催化性能[14-16],而載體、助劑的使用不僅提高了成本也增加了實驗的難度和不可控因素。
金屬?有機框架化合物(meta1-organic frame-works,MOFs)是以金屬離子或金屬簇作節點、以有機配體作連接體,通過兩者間某一種或多種作用力組裝成的周期性多孔結構[17]。這種結構可以將活性金屬均勻分散[18],焙燒后有效地防止了活性組分的燒結團聚,同時配體也會對雙活性位點的協同作用、催化劑表面酸堿性調節[19]產生一定作用。因此本課題組前期嘗試了引入MOFs結構來改善催化劑的穩定性和C2+醇產物選擇性,研究發現,配體在氮氣氣氛焙燒后可得到均勻分散的低價金屬氧化物或碳化物,一定程度上避免了活性金屬的燒結,但由于制備方法是將Cu浸漬到MIL-88B(Fe)結構中,沒有實現2種活性金屬均固定在MOFs骨架中。為了進一步將2種活性金屬同時固定在MOFs材料骨架,并引入堿性配體來提高CO2的活化性能,探索MOFs材料配體組成及比例對催化劑表面活性物種分布以及其在CO2加氫制C2+醇反應中的活性關聯和制約機制,我們以硝酸銅、硝酸鐵為金屬源,嘗試通過在MIL-88B(Fe)前驅體制備過程中引入第二種配體對氨基苯甲酸(p-ABA),并通過改變對苯二甲酸(PTA)和對氨基苯甲酸的比例來調控催化劑表面Fe物種的組成,進而考察表面組成與催化活性間的關聯因素。
1 實驗部分
1.1 實驗原料
硝酸銅(Cu(NO3)2·3H2O)、硝 酸 鐵 (Fe(NO3)3·9H2O)、無水氫氧化鈉(NaOH)、N,N-二甲基甲酰胺(DMF)購自天津市科密歐化學試劑有限公司;甲醇購自天津市匯杭化工科技有限公司;PTA、p-ABA購自美國Sigma-A1drich公司;蒸餾水(DI)購自太原理工大學中試基地(pH≈7);石英砂(SiO2)購自天津化學試劑三廠,以上試劑均為分析純。H2(純度不低于99.99%)、CO2(純度不低于 99.99%)、N2(純度不低于99.99%)購自太原市福江特種氣體有限公司。
1.2 儀器設備
合成中所用儀器有:FA2003型電子天平(天津天馬儀器廠)、DF-101S型磁力水浴鍋(天津工興電器廠)、BT100K型蠕動泵(河北保定創銳有限公司)、FYF2008型水熱釜(上海志澤生物科技發展有限公司)、101-1型電熱鼓風干燥箱(北京科偉永興儀器有限公司)、TG16-WS型臺式高速離心機(湖南湘儀實驗室儀器開發有限公司)、SK2-4-2Q型管式爐(天津市天有利電爐有限責任公司)、DF-4A型壓片機(天津港東科技發展有限公司)。
1.3 催化劑制備
將20.20 g Fe(NO3)3·9H2O、8.31 g PTA和一定量p-ABA分別溶于DMF溶液中,待分散均勻后將2種溶液混合攪拌30 min,用蠕動泵滴加到20 mL NaOH(4 mo1·L?1)溶液中,滴加完畢后溶液在室溫下攪拌30 min,然后轉移到100 mL內襯為聚四氟乙烯的反應釜中,100℃下水熱反應24 h。冷卻至室溫,去掉上層清液后,依次用DMF、甲醇溶液洗滌、離心3次。將固體分離物放置烘箱在60℃干燥12 h。將所得固體(Fe基MOFs)緩慢分散到250 mL Cu(NO3)2·3H2O的甲醇溶液(mCu(NO3)2?3H2O∶mFe-MOF=0.15)中,室溫下攪拌24 h,然后升溫至90℃攪拌蒸干,繼續于烘箱中60℃下干燥12 h后,在管式爐氮氣氣氛下650℃焙燒4 h,制得Cm∶n催化劑,m∶n為PTA和p-ABA的物質的量之比。
1.4 材料表征
X射線衍射(XRD)測試在DX-2700型X射線衍射儀(丹東方圓)上進行,采用Cu靶Kα射線(λ=0.154 184 nm),工作電流為30 mA,電壓40 kV,掃描范圍5°~85°,掃描速率8(°)·min?1。
H2程序升溫還原(H2-TPR)測試在TP-500型吸附儀(天津先權)上進行。將50 mg催化劑升溫至150℃,用氦氣吹掃30 min,再降溫至50℃,切換為體積分數為5% H2-95% N2的還原氣吸附30 min,然后以10℃·min?1速率升溫至800℃進行程序升溫還原,用熱導檢測器(TCD)檢驗耗氫量。
N2吸附?脫附表征在QDS-30型物理吸附儀(美國康塔)上進行。將催化劑造粒后在真空200℃下預處理4 h,然后在液氮中采用N2吸附法測定,比表面積采用 BET(Brunauer-Emmett-Te11er)法計算,孔容、孔徑采用 BJH(Barret-Joyner-Ha1enda)法計算。
掃描電子顯微鏡(SEM)測試在Quanta 400 FEG型場發射電子顯微鏡上進行,加速電壓為20 kV,采用能量分布面掃描分析(EDS-mapping)來分析元素的分散性。
X射線光電子能譜(XPS)測試在ESCALAB 250型光譜儀(賽默飛世爾科技公司)上進行,A1 Kα為輻射源(hν=1 486.6 eV,12.5 kV,16 mA),真空度為p=8×10?10Pa,以結合能為 284.8 eV 的 C1s為標準進行校正。
1.5 催化劑活性評價
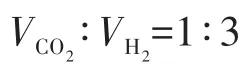
2 結果與討論
2.1 結構分析
圖1為制備的催化劑的XRD圖。由圖1a可知反應前所有催化劑均出現Fe0(PDF No.87-0721)、Cu0(PDF No.02-1225)、Fe3O4(PDF No.89-2355)、FeO(PDF No.89-0687)的特征衍射峰,但其結晶度存在差異;結合圖1b發現隨著p-ABA比例的增大,Fe3O4衍射峰減弱,FeO衍射峰增強,說明p-ABA的加入有利于低價態鐵的形成;由圖1c可知反應后催化劑物相除Cu0、Fe3O4外,在 2θ=41.2°、44.1°、47.3°位置處出現Fe5C2(PDF No.51-0997)物種的特征衍射峰;2θ=38.2°、41.1°、43.6°處出現Fe3N(PDF No.83-0877)物種的特征衍射峰,說明催化劑在反應過程中Fe物相發生了變化[20]。比例為5∶2時低價 Fe3O4、Fe5C2物種的衍射峰最強,說明這種狀態下低價態鐵含量最多。
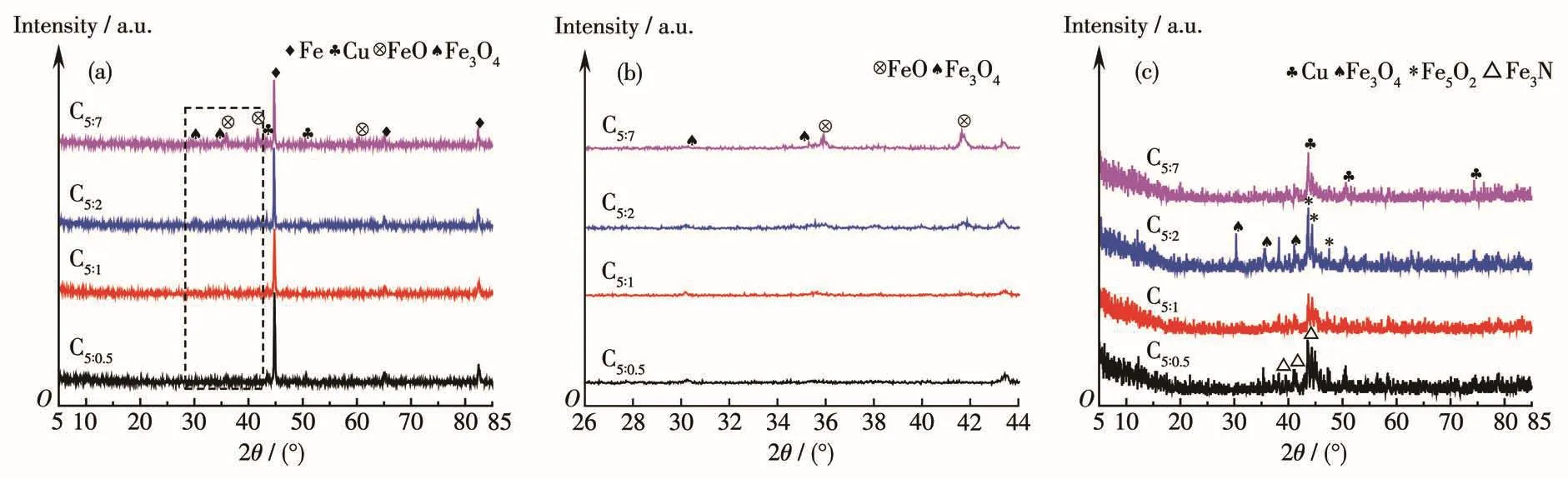
圖1 制備的催化劑的XRD圖:(a)反應前;(b)局部放大;(c)反應后Fig.1 XRD patterns of as-prepared cata1ysts:(a)before reaction;(b)partia1 en1arged drawing;(c)after reaction
根據Scherrer公式計算了催化劑反應前后的Cu0晶粒尺寸(表1)。由表可知,反應后顆粒尺寸發生不同程度的減小,說明催化劑結構對Cu物種有較好的分散性,反應過程中沒有發生團聚,而小尺寸銅晶粒擁有比較多的開放位面和邊緣缺陷位點,有利于和關鍵反應中間產物的結合[21],促進反應的進行。
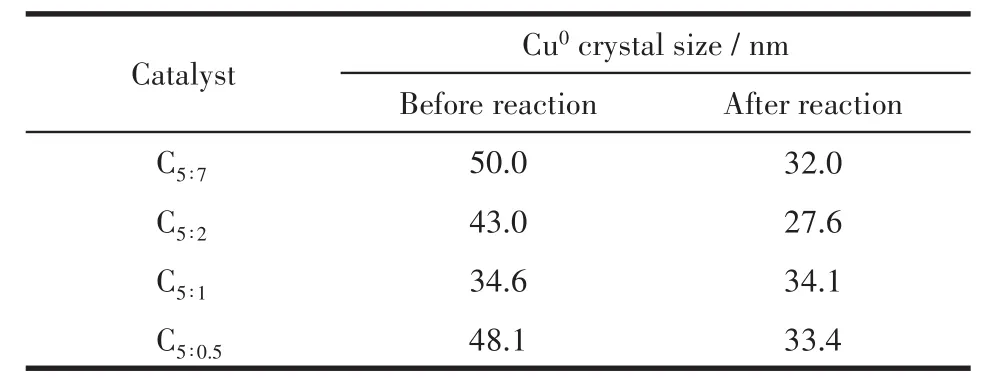
表1 制備的催化劑中Cu0晶粒尺寸Table 1 Crystal size of Cu0in as-prepared catalysts
2.2 N2吸附-脫附分析
圖2為制備的催化劑反應前(a、b)后(c、d)的N2吸附?脫附等溫線和孔徑分布圖。根據IUPAC分類,反應前后催化劑均為Ⅳ型等溫線,說明催化劑在反應過程中結構沒有發生變化,屬于典型的介孔材料。低壓區(p/p0<0.4)樣品的N2吸附量逐漸增加,屬于物理吸附;高壓區(p/p0>0.4)出現H3型回滯環,說明沒有出現限制性吸附[23]。由孔徑分布圖可知反應前后催化劑孔結構豐富,2~32 nm范圍的孔同時存在,2~7 nm孔分布較均勻,3~4 nm孔最多,并且p-ABA用量的改變不會對其結構產生明顯影響。
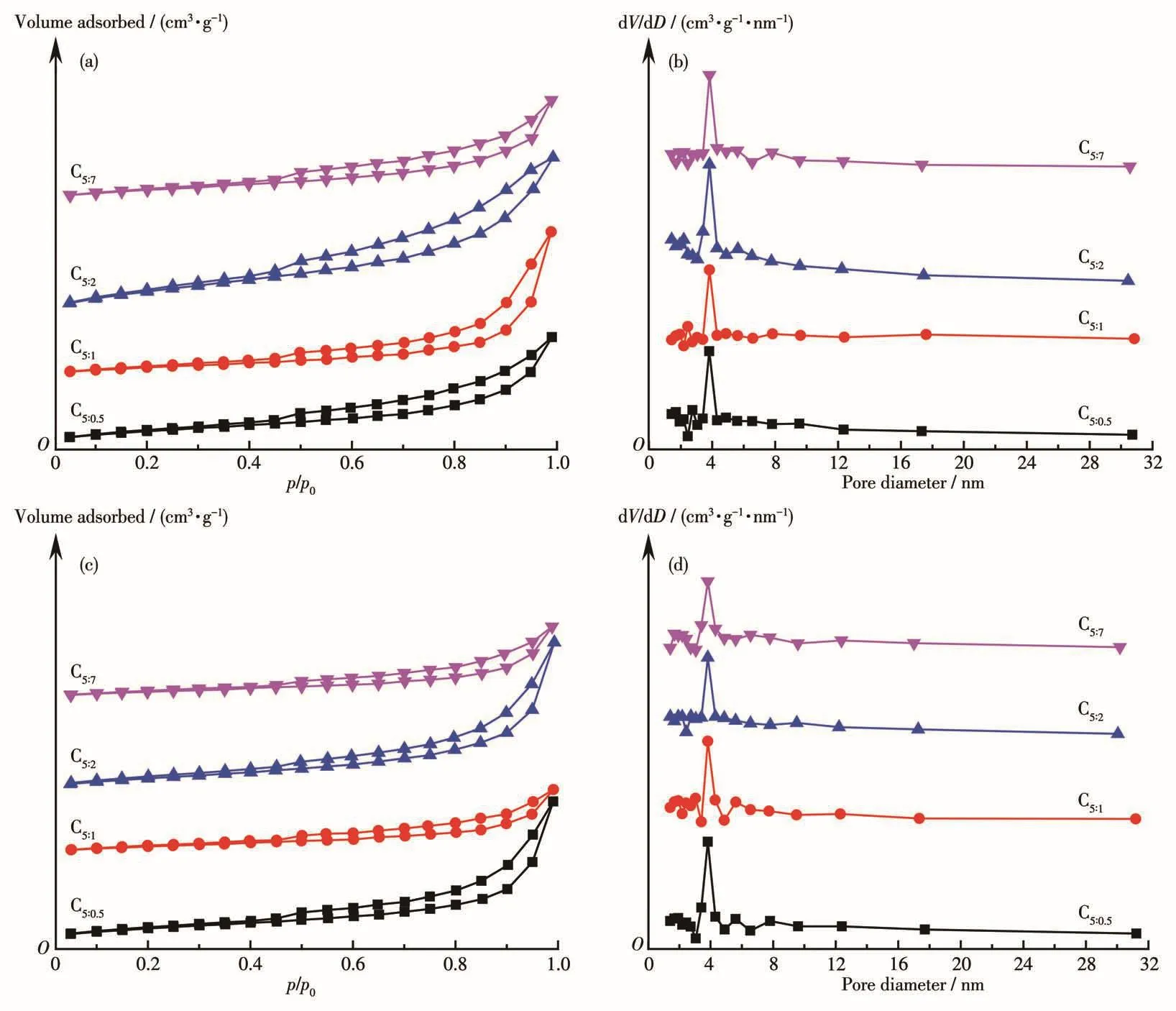
圖2 制備的催化劑反應前(a、b)后(c、d)的N2吸附?脫附等溫線和孔徑分布圖Fig.2 N2-adsorption-desorption isotherms and pore size distribution curves of as-prepared cata1ysts before(a,b)and after(c,d)reaction
表2列出了催化劑反應前后的織構參數,隨著p-ABA的增加,反應前(后)各催化劑之間比表面積、孔容、孔徑差異較小,說明p-ABA用量對催化劑結構性質影響較小;相比于反應前,反應后催化劑比表面積發生不同程度的減小,孔容、孔徑變化較不明顯,可能是反應過程中輕微積碳所致。C5∶2催化劑反應后比表面積、孔容偏大,晶粒尺寸偏小,結合SEM、EDS-Mapping分析結果可推測,此催化劑中活性組分分散均勻,沒有嚴重的燒結、團聚現象發生,結合活性數據,這可能是其催化活性較高的原因之一。
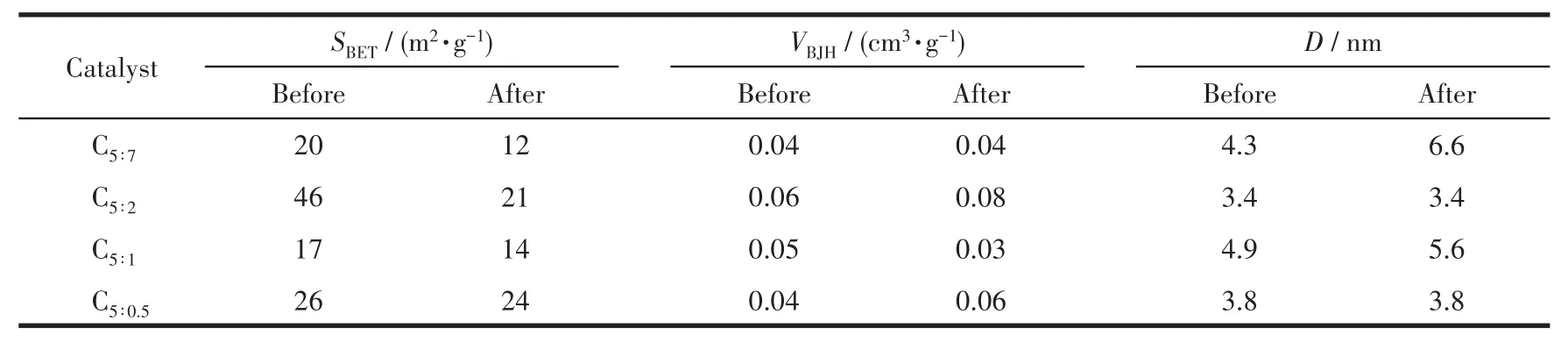
表2 制備的催化劑反應前后的織構性質Table 2 Texture parameters of as-prepared catalysts before and after reaction
2.3 H2-TPR分析
為了進一步研究催化劑原位活化后的活性物種組成,對焙燒后催化劑進行了H2-TPR表征(圖3)。可見催化劑均在190~250℃和250~650℃范圍內出現2個主要的還原峰,分別歸屬于Cu物種(Cu2+→Cu0)、Fe物 種 (Fe3O4→FeO→Fe)的 還 原[20,22],隨 著p-ABA比例的增大,Cu物種還原峰向低溫發生偏移,Fe物種還原峰向高溫發生移動。第一個峰的出現說明催化劑中還存在銅的氧化物,由于其含量低或者分散性較好,XRD中沒有出現對應的特征衍射峰;目前關于Fe3O4、FeO的還原溫度沒有明確的界定,所以第二個峰歸屬于低價態鐵的還原。H2-TPR、XRD表征結果結合二氧化碳加氫反應條件可知,催化劑在反應前經氫、氮混合氣350℃還原后,真正的活性組分為Cu0、Fe3O4以及以低價態形式存在的Fe物種。
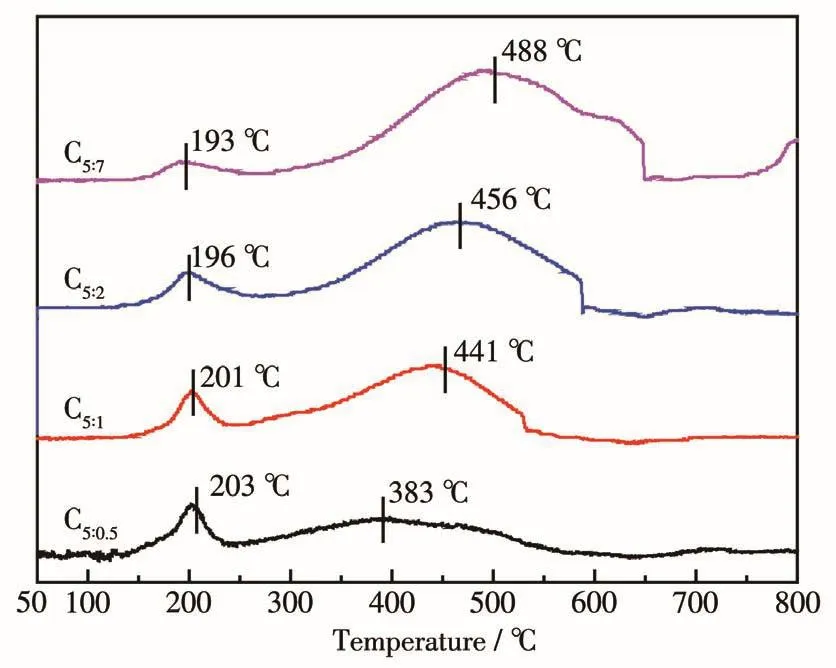
圖3 制備的催化劑的H2-TPR曲線Fig.3 H2-TPR profi1es of as-prepared cata1ysts
2.4 SEM分析
為了清晰地看到催化劑表面形貌及各元素分布,對 C5∶2催化劑(反應后)做了 SEM 和 EDS-Mapping表征。如圖4所示,從圖a中可以看到催化劑出現均勻的片層狀結構,堆疊形成豐富的孔道,結合圖2b孔徑分布結果可知,這可能是催化劑同時存在2~30 nm尺寸范圍孔道的原因。增大放大倍數(圖4b和4c),出現梭形桿狀結構,這些結構更有利于活性位點的暴露,促進催化劑與原料氣體充分接觸。圖4e(Cu)、4f(Fe)、4g(O)為 EDS-mapping元素分布圖,可以看出Cu、Fe、O元素均勻分散,進一步證明了配體的引入有利于Cu、Fe組分的分散和相互作用,同時也有利于活性組分的暴露,增大與反應氣的接觸幾率,促進催化反應的進行。

圖4 催化劑C5∶2的SEM圖 (a~d)和Cu、Fe、O元素分布 (e~g)Fig.4 SEM images(a~d)and e1ement distribution(e~g)of C5∶2cata1yst
2.5 XPS分析
應用XPS對催化劑反應后的組成、金屬價態進行了分析。表3為催化劑各元素表面含量,除C、O、Cu、Fe主要元素外,還有微量N元素,與XRD表征結果中Fe3N物種衍射峰的出現一致。由于該系列催化劑在制備過程中配體用量較多,熱解條件下分解產生的C富集于表面,導致其原子占比多達52.38%以上;隨著p-ABA用量的增加,Cu、Fe金屬原子占比呈現先增大后減小的火山型趨勢,配體比例為5∶2時Cu、Fe位點暴露最多,分別為2.38%、3.29%,這也是C5∶2催化劑活性較好的原因之一;N元素含量隨p-ABA 比例增大逐漸增多,nPTA∶np-ABA=5∶2 時達到0.77%,進一步增大用量其N元素含量變化較不明顯,從活性金屬表面含量和經濟成本兩方面考慮,配體PTA、p-ABA的最佳比例為5∶2。有文獻指出Fe0[24]、Fe5C2[25-26]有利于碳鏈增長,Fe2+有利于 H2解離,為了探究催化劑表面Fe活性物種的存在形式,關聯配體組成及配比與活性物種分布及催化劑活性間的關系,圖5對反應后Fe2p XPS(a)及其分峰擬合譜圖(b)進行了分析。圖5a中該系列催化劑在結合能為724.4、711.2 eV處出現2個峰,并且其自旋軌道耦合(ΔE)為13.2 eV,這些特征參數對應于Fe3O4物種中的Fe3+和Fe2+[27];在718.2 eV附近沒有出現衛星峰,說明反應后催化劑中不存在Fe2O3[28],與 XRD表征結果相一致,表明Fe被還原為低價態物種。為了計算不同形式的Fe含量,對Fe2p進行了分峰擬合,結合能712.5、710.7、707.5 eV處的峰分別歸屬于Fe3+、Fe2+、Fe0或Fe5C2[29-31],從計算結果(表 4)可以發現隨著p-ABA用量的增加,Fe3+占比先減小后增大,Fe2+及低價態鐵(Fe2+、Fe0和 Fe5C2總和)占比先增大后減小,nPTA∶np-ABA=5∶2 時低價態鐵的原子占比最大,為71.27%,說明催化劑C5∶2中Fe主要以低價形式存在,與XRD表征結果(圖1b)一致。結合活性數據發現,低價態Fe含量高有利于醇類尤其是C2+醇的生成,這與本課題組前期的研究結果相一致[32]。同時通過對C1s XPS譜圖(圖6)的分析發現,反應后催化劑均出現4個峰,結合能位于289.5 eV的峰歸屬于C=O鍵,285.6 eV處的峰歸屬于C—C鍵,低結合能284.8、284.1 eV處的峰分別歸屬于表面無定形碳物種和 FeCx物種[33-34],結合 XRD、Fe2p XPS 結果可知,FeCx物種在催化劑中主要以Fe5C2形式存在。
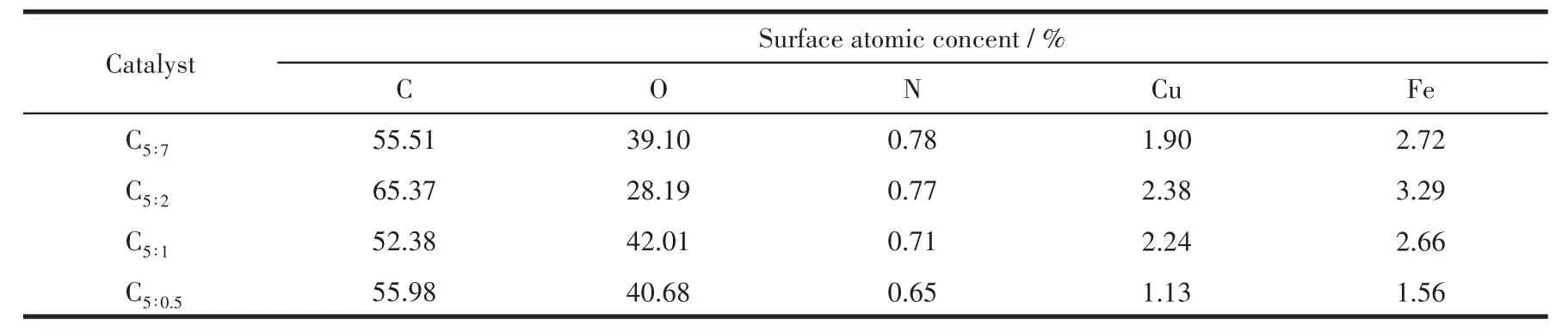
表3 基于XPS的所制備催化劑中各元素組成及含量Table 3 Elemental analysis results of as-prepared catalysts derived from XPS
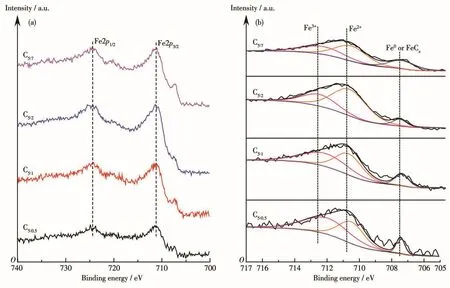
圖5 制備的催化劑反應后的Fe2p XPS譜圖Fig.5 Fe2p XPS spectra of as-prepared cata1ysts after reaction
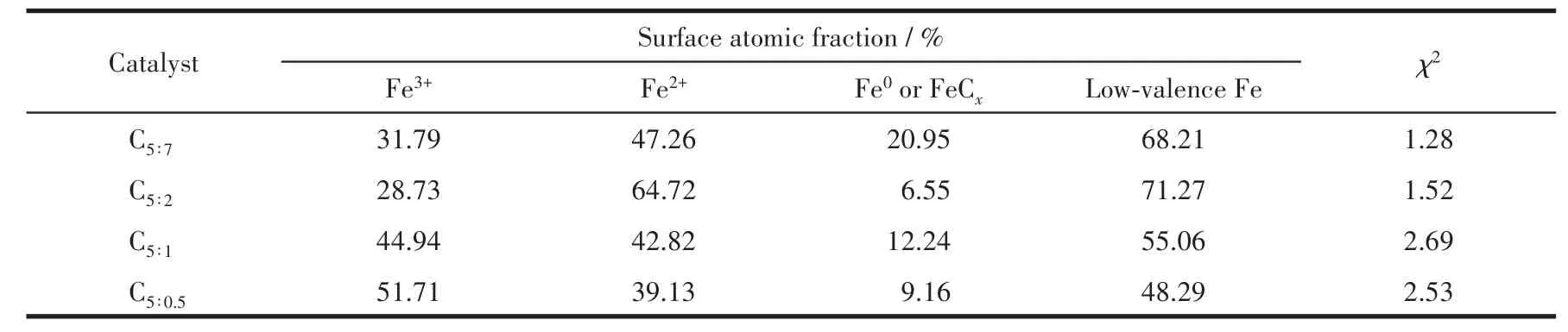
表4 制備的催化劑反應后樣品表面Fe物種組成及占比Table 4 Composition and proportion of Fe species in the surface of as-prepared catalysts after reaction
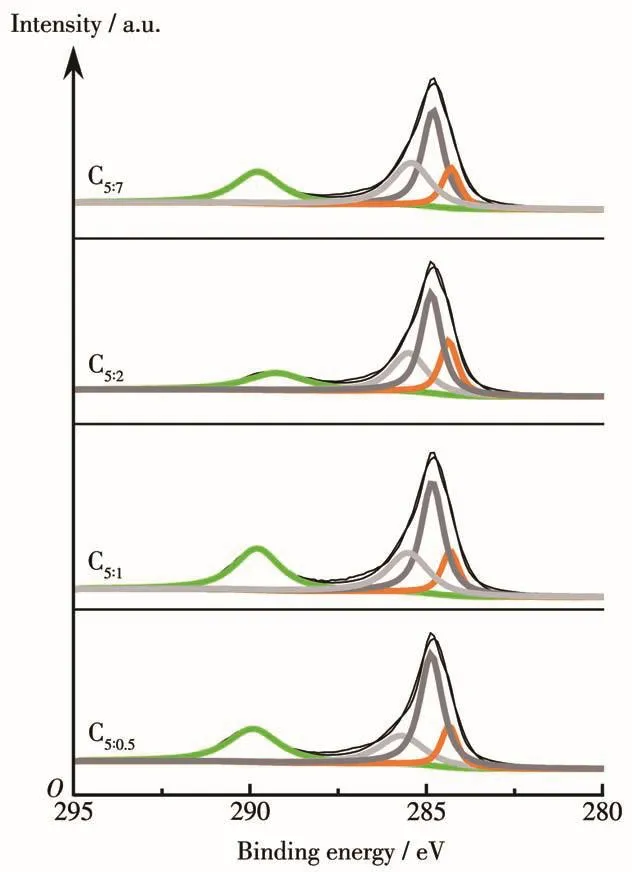
圖6 制備的催化劑反應后的C1s XPS譜圖Fig.6 C1s XPS spectra of as-prepared cata1ysts after reaction
2.6 催化劑活性分析
表5列出了制備的催化劑用于CO2加氫反應的活性評價數據。可見,反應產物以CO、烴類為主,約占總產物的70.0%。隨著p-ABA用量的增加,CO2轉化率不斷增大,說明氮摻雜有利于促進CO2的吸附、活化,可能是由于摻雜的N會與活性金屬、CO2發生相互作用,從而降低CO2解離的活化能[35-36];同時CO選擇性先減小后增大,總醇選擇性先增大后減小,與低價態鐵的含量的變化趨勢(表4)一致,當配體PTA和p-ABA的比為5∶2時,催化劑表面低價態鐵含量最高,總醇(ROH)選擇性最大,CO選擇性最小,說明低價態鐵的存在能夠抑制RWGS反應,有利于反應向生成醇的方向進行,此時C2+醇(C2+OH)的時空收率(STY)達到峰值45.49 mg·mL?1·h?1。
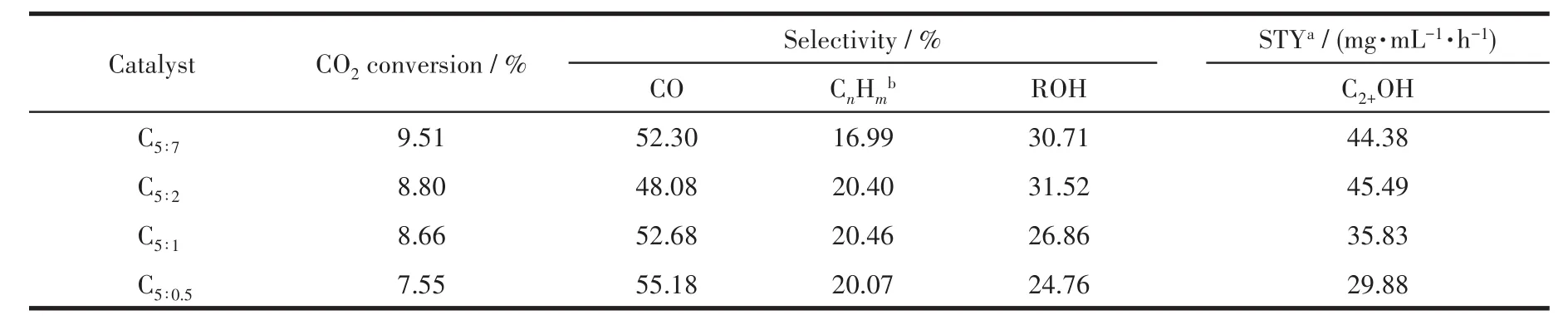
表5 制備的催化劑的催化性能Table 5 Catalytic performance of as-prepared catalysts
表6展示了產物中各種醇的選擇性及在總醇中的分布。由表可以看出,隨著p-ABA用量的增加,甲醇、丙醇選擇性相對較小且呈現波動式變化,說明氮含量的變化對其沒有明顯作用規律;而此時乙醇選擇性先增大后減小,與總醇選擇性變化趨勢保持一致。根據各類醇在總醇中的分布可知,產物醇中主要以乙醇為主,可能歸因于催化劑結構的限域作用,當最可幾孔徑為3~4 nm時,乙醇的物質的量分數最高,而較大的孔徑則傾向于生成乙醇偶聯產物丁醇。隨著p-ABA用量的增加,丁醇選擇性明顯變大,當2種配體的比例為5∶7時最大,此時Fe5C2占比最多,Fe5C2被認為能促進C—C偶聯,有利于碳鏈增長[25,37-38],這可能是丁醇選擇性增大的原因。
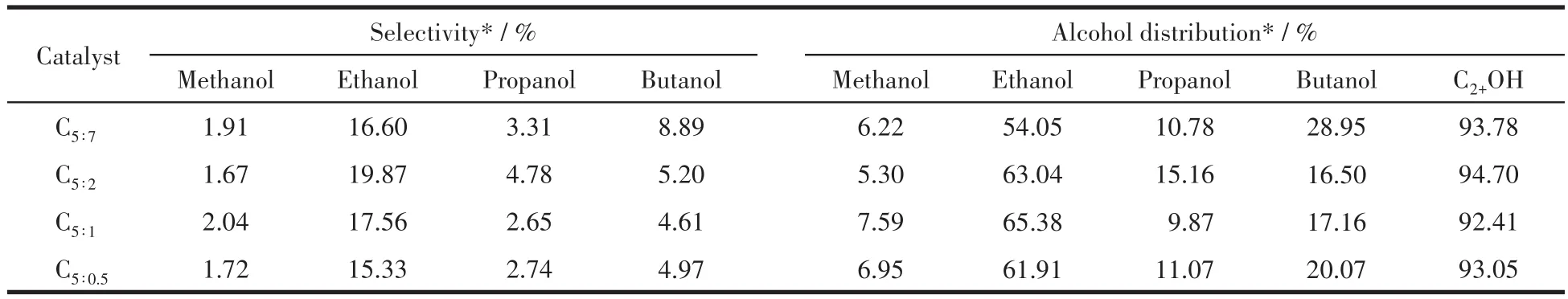
表6 制備的催化劑催化作用下醇產物中各類醇分布Table 6 Alcohol distributions in total alcohol catalyzed by as-prepared catalysts
在總醇中,甲醇的物質的量分數較低,C2+醇總物質的量分數都達到了92.0%以上,其中乙醇占主要部分。圖7對低價態鐵占比與總醇選擇性(a)、低價態鐵占比與C2+醇選擇性(b)進行了線性擬合,R2分別達到0.999 8、0.998 8,呈現較好的相關性,說明低價態鐵有利于CO2加氫合成C2+醇,關于其反應機理有待進一步研究。綜上所述,分散性較優的催化劑結構有利于低價態鐵的形成,進而促進了CO2加氫制C2+醇,2種配體比例為5∶2時低價態鐵占比最高,催化效果最佳,CO2轉化率為8.80%,總醇選擇性為31.52%,其中C2+醇物質的量分數達到94.70%。表7對不同CuFe基催化劑用于CO2加氫制C2+醇的催化性能進行了對比,可以發現與多組分催化劑相比,以MOFs為前驅體制備的CuFe雙組分催化劑用于CO2加氫制C2+醇時,催化性能較優,反應條件相對溫和,GHSV較高,產物醇中C2+醇物質的量分數較高。
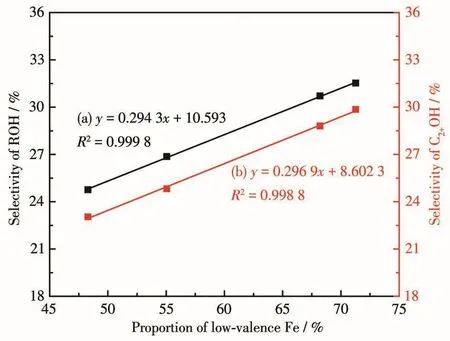
圖7 低價態鐵含量與總醇選擇性(a)及C2+醇選擇性(b)關系圖Fig.7 Re1ationships between content of 1ow-va1ence iron and se1ectivity of ROH(a)and C2+OH(b)
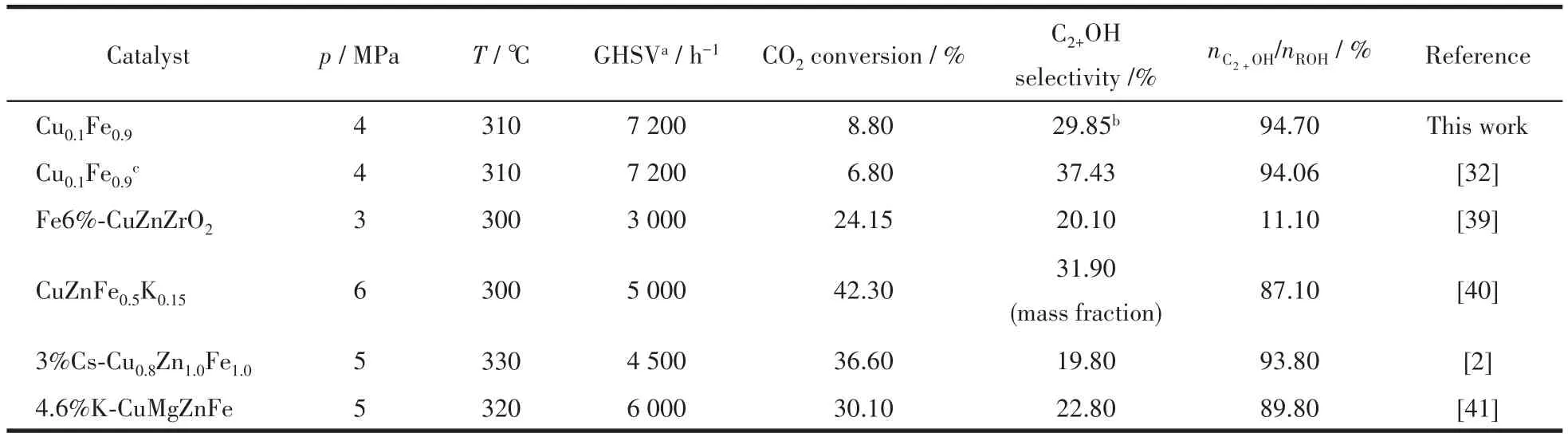
表7 CuFe催化劑用于CO2加氫制C2+醇的催化性能Table 7 Catalytic performance of CuFe catalyst for CO2hydrogenation to C2+alcohol
3 結論
以雙配體CuFe@MOFs材料為前驅體制備的催化劑用于CO2加氫制C2+醇,通過實驗研究得出以下結論:
(1)SEM、EDS-mapping表征發現,采用水熱浸漬法、氮氣焙燒氣氛制備的CuFe基催化劑活性組分分散均勻,避免了高溫加氫條件下Cu組分團聚、燒結現象的發生。
(2)由XPS數據可知,調節2種配體(對苯二甲酸、對氨基苯甲酸)的比例可以調控催化劑中低價態鐵的分布,進而對CO2加氫合成C2+醇的催化性能產生影響。當對苯二甲酸和對氨基苯甲酸比例為5∶2時,催化劑中低價態鐵占比最高,對C2+醇的生成最有利。
(3)銅鐵組分良好的分散性是合成醇的關鍵,低價態鐵的存在對反應產物的碳鏈增長起重要作用。
猜你喜歡
課堂內外·初中版(科學少年)(2025年1期)2025-02-28 00:00:00
課堂內外·初中版(科學少年)(2025年2期)2025-02-28 00:00:00
英語世界(2023年10期)2023-11-17 09:18:18
科學大眾(中學)(2019年3期)2019-05-17 10:04:30
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
汽車觀察(2018年10期)2018-11-06 07:05:26
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
中國資源綜合利用(2016年4期)2016-01-22 08:27:23

- 無機化學學報的其它文章
- 更正:基于2,6-二(4-羧基苯亞甲基)環己酮的金屬-有機框架化合物的合成與表征
- Improving Energy Barrier by Altering Coordination Environment in Two Dy(Ⅲ)Single-Ion Magnets
- Two-Dimensional Luminescent Coordination Polymer Based on Dinuclear{Zn2(COO)4}Second Bulidings Units:Crystal Structure and Detection of Fe3+
- Syntheses,Spectroscopic Properties and Terahertz Time Domain Spectroscopy of Two Copper(Ⅰ)Complexes Based on Diphosphine Ligands and N-Donor Ligands
- Palladium-Based Coordination“Clips”with Carboxamide-Pyrazolate Ditopic Ligands:Self-Assembly and Catalytic Properties
- Synthesis of High-Flux Mordenite Membranes by Binary Cations System for Pervaporation Dehydration of Acetic Acid