穩定同位素氘標記的去甲烏藥堿的合成與表征
2021-08-19 04:24:36韓世磊陳菊玲楊立鳳
同位素 2021年4期
韓世磊,徐 銀,陳菊玲,楊立鳳,張 磊
1.天津阿爾塔科技有限公司,天津 300457; 2.阿爾塔(天津)標準物質研究院有限公司,天津 300457)
去甲烏藥堿最初是由Kosuge等從日本附子中分離提取,在臨床上用作強心劑、利尿劑、鎮痛劑和降壓劑等,服用或者誤服將會對運動員的成績造成較大影響[1-2]。世界反興奮劑機構(world anti-Doping agency, WADA)在《2019年禁用清單國際標準》中已將其明確列為β2激動劑類禁用物質。2019年8月中國體育總局反興奮劑中心關于印發《大型賽事食源性興奮劑》防控工作指南(暫行)的通知中,明確規定要檢測沙丁胺醇、萊克多巴胺、沙美特羅、克倫丙羅、去甲烏藥堿和曲托奎酚等β2激動劑。因此,有效的檢測手段運用于各種食材的檢測以及人體代謝物的檢測尤為重要。日前,關于去甲烏藥堿的檢測方法主要有高效液相色譜-紫外檢測(HPLC-UV)法、液相色譜-熒光檢測(HPLC-FLD)法、高效液相色譜-電化學檢測(HPLC-CECD)法和液相色譜-質譜(LC-MS)法等[3-6],但是其靈敏度有一定的局限性,當濃度低于最低定量限時就無法滿足要求。同位素稀釋質譜法(isotope dilution mass spectrometry, IDMS)采用穩定性同位素標記化合物作為內標試劑,結合了色譜的分離能力和質譜的定性能力,通過檢測相應質量數的離子比值與標準比值可準確定量,同時消除樣品前處理過程中引起的基質效應和回收率差異,提高檢測準確度[8]。因此,穩定同位素內標試劑是該檢測方法能否使用的關鍵因素。
當前,去甲烏藥堿已經可以化學全合成,技術日趨成熟,諸多文獻已對其原藥的合成進行了相關報道[9-11],但關于去甲烏藥堿的同位素標記化合物的文獻報道較少。有文獻報道了氚(T)標記去甲烏藥堿化合物和去甲烏藥堿-13C標記化合物的生物合成方法,但是放射性及實用性問題使其應用受到限制[12-13]。此外,有文獻報道了一種關于中間體2-(3,4-二甲氧基苯)乙胺-D4的合成方法[15],其以2-(3,4-二甲氧基苯)乙腈為原料,以雷尼鎳為催化劑,以D2為氘源,借助流體化學技術,合成目標產物,收率達70%,但是與氨基相鄰碳上的氘代交換率僅17%,無法滿足要求。同時,該方法需要使用特殊的設備以及有安全風險的氘氣,因此并不適合普通實驗室制備。為解決內標試劑的來源問題,本研究擬開發一條收率高、操作簡便、同位素豐度高的合成路線,以期為更準確地定量檢測去甲烏藥堿的痕量殘留提供標準內標試劑。
1 實驗部分
1.1 主要儀器
IKA-RCT basic型加熱型磁力攪拌器:德國IKA集團;IKA HB10型旋轉蒸發儀:德國IKA集團;Bruker 600 MHz型核磁共振色譜儀:德國布魯克公司;Agilent1260+6120型高效液相色譜-質譜聯用儀:配自動進樣器與DAD檢測器,美國安捷倫科技公司;CHEETAH MP200型快速柱純化儀:天津博納艾杰爾科技有限公司;LC-IT-TOF-MS型高分辨質譜儀:日本島津公司。
1.2 主要材料與試劑
2-(3,4-二甲氧基苯)乙腈:薩恩化學技術(上海)有限公司;重水(99.8%D)、氘代硼氫化鈉(98%D)、氘代氫化鋁鋰(98%D):美國劍橋穩定同位素公司;4-甲氧基苯乙醛:上海畢德醫藥;其他試劑與藥品均為市售分析純,除特別說明外,未經處理直接使用。
1.3 實驗方法
穩定同位素氘標記的去甲烏藥堿(6)的合成,以2-(3,4-二甲氧基苯)乙腈為起始原料,經過堿催化的氫-氘同位素交換、氘還原、皮克特-施彭格勒(Pictet-Spengler)環化反應、脫甲基等關鍵反應步驟,得到四氘代的去甲烏藥堿。合成路線示于圖1。
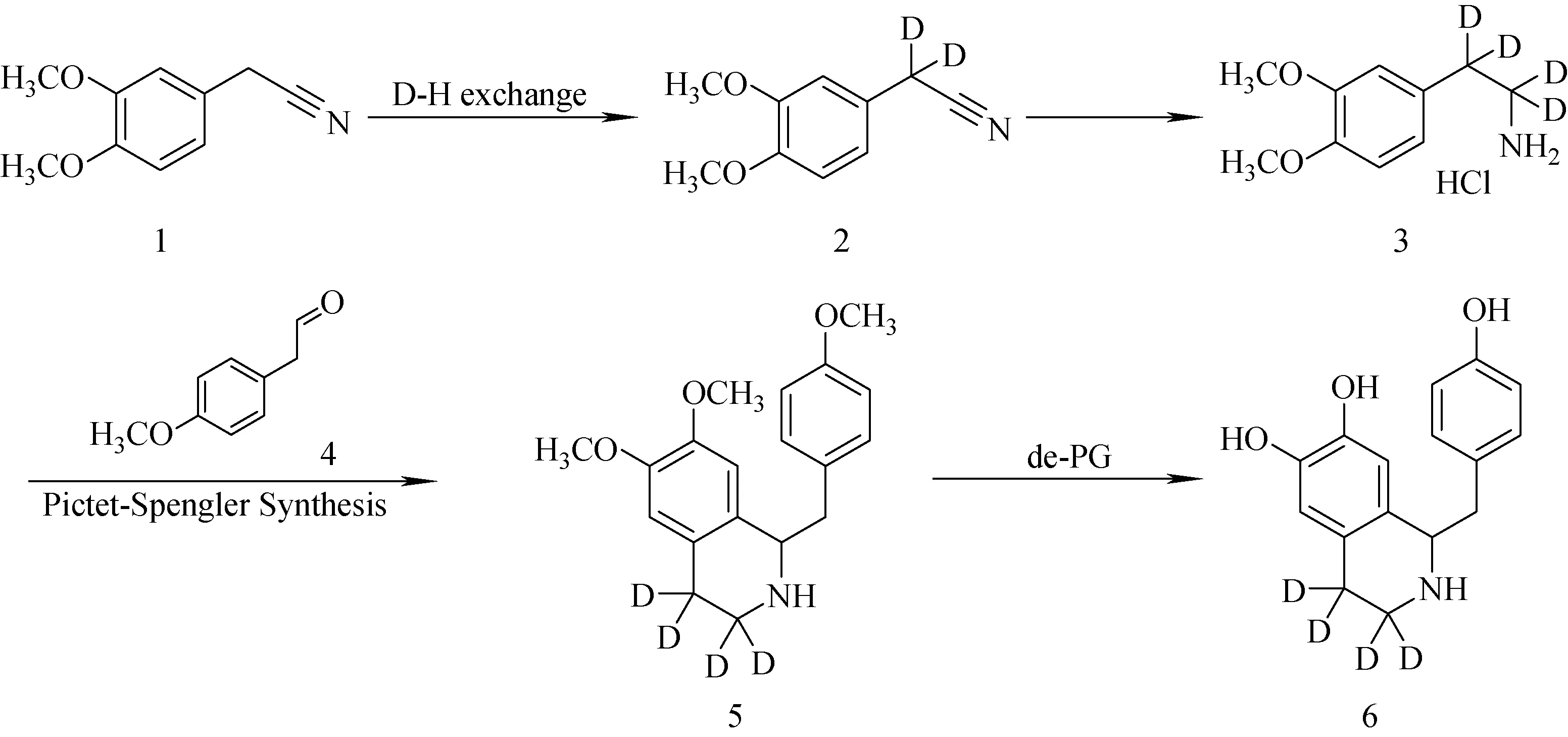
圖1 去甲烏藥堿-D4的合成路線Fig.1 Scheme for the synthesis of higenamine-D4
1.3.12-(3,4-二甲氧基苯)乙腈-D2(2)的合成 于氮氣保護下將3,4-二甲氧基苯乙腈(1)(10.0 g)溶于60 mL的干燥THF和100 mL的D2O中,加入預先干燥的K2CO3(31.0 g, 4 eq,eq表示物料當量),反應混合物于室溫攪拌48 h。加入干燥的叔丁基甲基醚萃取,分液,有機相干燥、過濾,旋轉蒸發濃縮后真空干燥得到2-(3,4-二甲氧基苯)乙腈-D2(2)(9.5 g),收率95.0%。1H NMR(600 MHz,Chloroform-d)δ6.87-6.82(m,2H),6.80(d,J=1.9 Hz,1H),3.88(d,J=8.5 Hz,6H)。
1.3.22-(3,4-二甲氧基)苯乙胺鹽酸鹽-D4(3)的合成 于氮氣保護下將化合物(2)(3.0 g)溶于干燥的THF和MeOD的混合溶劑(5∶1,60 mL)中,依次加入(Boc)2O(8.0 g,2.0 eq)、氯化鎳(0.23 g,1.1 eq),冰浴冷卻至10 ℃左右。攪拌下分批加入氘代硼氫化鈉(1.29 g,2.0 eq),然后自然升至室溫,攪拌過夜。反應完畢后,加入飽和氯化銨水溶液淬滅,乙酸乙酯萃取,干燥、過濾,減壓濃縮,粗品經Flash柱色譜純化得到Boc保護的中間體,然后再加入1 mol/L的鹽酸/乙酸乙酯溶液(50 mL),室溫攪拌反應過夜。旋轉蒸發濃縮后真空干燥得到2-(3,4-二甲氧基苯)乙胺鹽酸鹽-D4(3)3.8 g,收率80.0%。1H NMR(600 MHz,DMSO-d6)δ8.03(br,3H),6.93-6.83(m,2H),6.75(dd,J=8.1,2.0 Hz,1H),3.74(d,J=19.2 Hz,6H)。
1.3.36,7-二甲氧基-1-(4-甲氧基苯基)-1,2,3,4-四氫異喹啉-3,3,4,4-D4(5)的合成 于氮氣保護下將化合物(3)(500 mg,1 eq)和對甲氧基苯乙醛(4)(500 mg,1.5 eq)溶于無水甲苯(20 mL)中,加入對甲苯磺酸(600 mg,1.5 eq),反應混合物加熱回流3 h。反應液冷卻后,用水洗滌,干燥、過濾、濃縮。粗產品經Flash柱色譜純化得到中間體(5)400 mg,收率55.0%。1H NMR(600 MHz,Methanol-d4)δ8.50(s,1H),7.26-7.19(m,2H),6.97(d,J=8.7 Hz,1H),6.82(s,1H),6.49(s,1H),4.67(t,J=7.4 Hz,1H),3.83(d,J=11.7 Hz,6H),3.66(s,3H),3.37(dd,J=14.1,7.1 Hz,1H),3.14(dd,J=14.1,7.7 Hz,1H)。
1.3.4穩定同位素氘標記的去甲烏藥堿(6)的合成 于氮氣保護下將化合物(5)(200 mg)溶于無水二氯甲烷中,冷卻至0 ℃。攪拌下緩慢滴加三溴化硼的二氯甲烷溶液(1 mol/L,3.8 mL,6 eq),加畢,升至室溫反應3 h。反應完畢后,用氫氧化鈉水溶液調節pH至12,分出水層,用鹽酸調節pH至7,析出的固體經過濾、干燥,粗產物經反相色譜純化后得到去甲烏藥堿-D4,89.2 mg,白色固體,收率51.4%,HPLC純度98.0%,氘豐度為96.5%。HRMSm/z:[M+H]+=276.152 8(理論值276.153)。1H NMR(600 MHz,Methanol-d4)δ7.14(d,J=8.5 Hz,1H),6.81(d,J=8.5 Hz,1H),6.63(s,1H),6.62(s,1H),4.58(dd,J= 8.9,5.6 Hz,1H),3.37(dd,J=14.6,5.6 Hz,1H),2.97(dd,J=14.6,9.0 Hz,1H)。
2 結果與討論
2.1 去甲烏藥堿-D4(6)的合成
2.1.12-(3,4-二甲氧基苯)乙腈-D2(2)的合成 在化合物(2)的合成研究中,對不同的氘源及堿作為氫-氘交換的反應條件進行優化,結果列于表1。從表1中可以看到,無論是有機金屬堿還是無機堿,底物交換反應后的同位素豐度均較高,但是有機金屬試劑作為堿時,反應體系有較多的雜質,收率相對較低,而用氫氧化鈉或者氘氧化鈉作堿時,產物收率明顯降低,推測可能是由于底物分子中的氰基水解所致,綜合實驗操作的簡便性及原輔材料廉價易得性,最終選擇K2CO3/THF/D2O體系進行該步氘-氫交換反應。
此外,針對表1中反應條件3的堿和重水的使用量、反應溫度及反應時間對反應收率和同位素豐度的影響進行研究,結果列于表2。由表2結果可知,反應溫度對同位素豐度影響較小,但是在較高溫度下反應收率有降低的趨勢,而增加重水的使用量和反應時間有利于提高產物的同位素豐度。
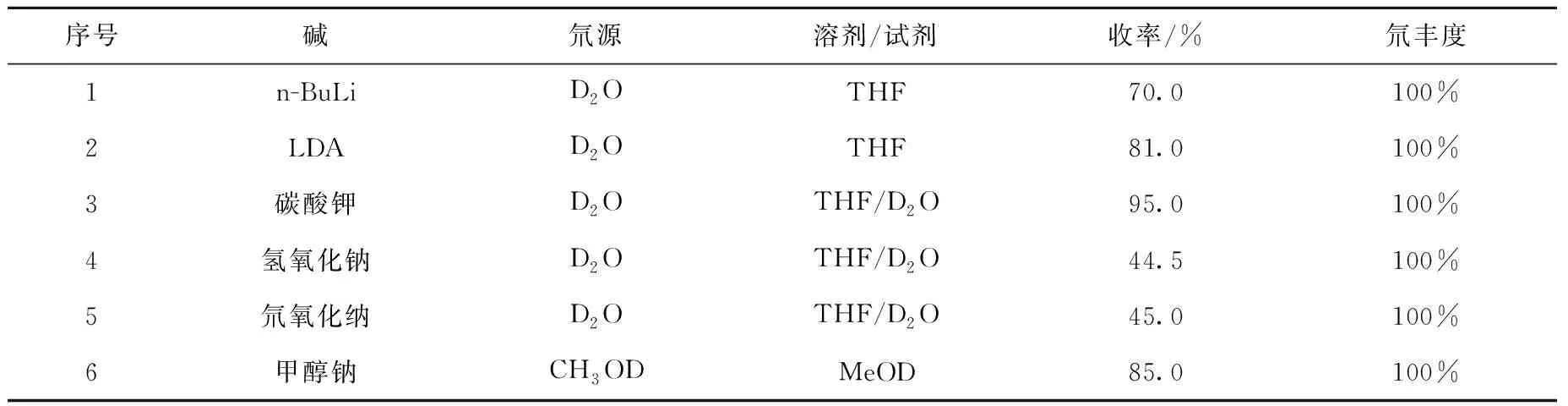
表1 化合物(2)合成的反應條件篩選Table 1 Reaction condition screening for the synthesis of compound (2)
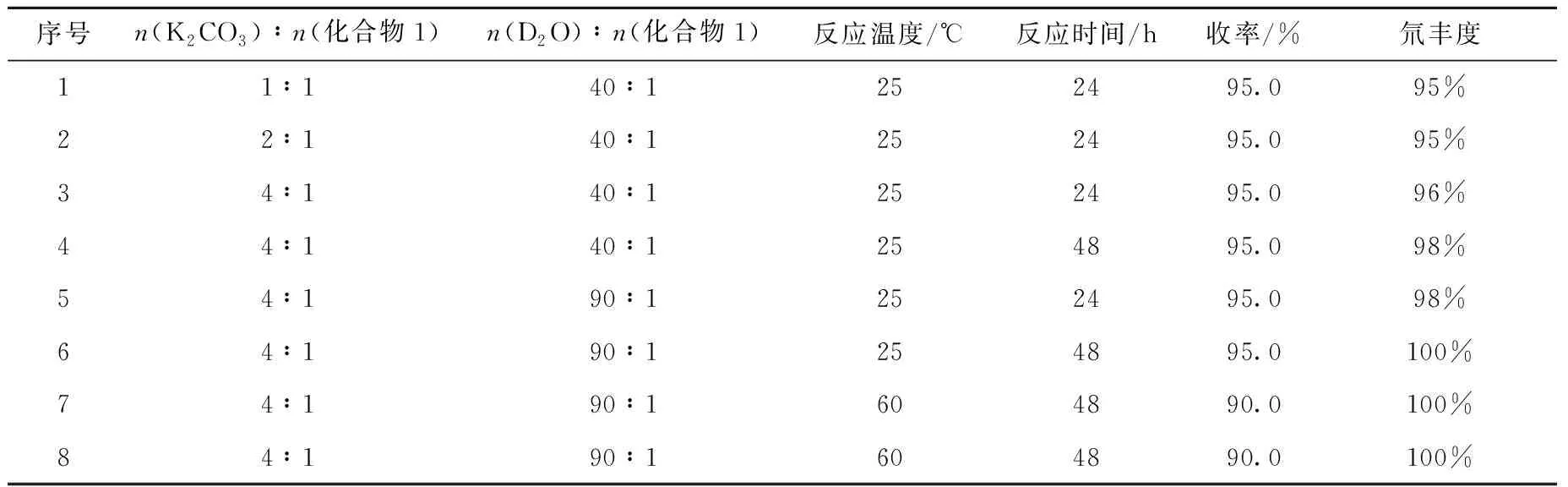
表2 試劑與底物的物質的量比對化合物(2)合成的影響Table 2 Effect of materials ratio on for the synthesis of compound (2)
2.1.22-(3,4-二甲氧基)苯乙胺鹽酸鹽-D4(3)的合成 在化合物(3)的合成研究中,針對不同還原劑、溶劑對反應的影響進行研究,結果列于表3。使用四氫鋁鋰作還原劑時,由于淬滅過程中產生的鋁鹽對產物的包裹,導致收率較低;而硼氫化鈉作為還原劑并進行“一鍋法”投料,使還原生成的胺原位生成Boc保護的胺,然后脫去Boc保護基得到目標化合物(3),可極大地提高產物收率。此外,使用四氫呋喃-甲醇做共溶劑可極大地提高反應速率,而用氘代甲醇(CH3OD)替換甲醇(CH3OH),可有效避免氫-氘交換,將產物的同位素豐度提高約97%。

表3 不同還原劑對化合物(3)合成的影響Table 3 Effect of reducing agents on the synthesis of compound (3)
2.1.36,7-二甲氧基-1-(4-甲氧基苯基)-1,2,3,4-四氫異喹啉-3,3,4,4-D4(5)的合成 化合物(5)的合成屬于Pictet-Spengler環化反應,本文對不同的酸性試劑及溶劑對反應的影響進行研究,結果列于表4。對甲苯磺酸作為質子酸體系催化效果較好,而其他酸性試劑如鹽酸體系結果不理想。
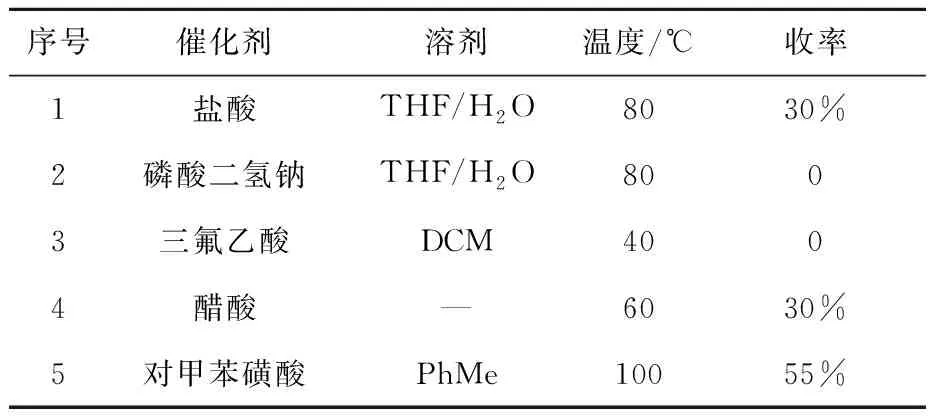
表4 催化劑對化合物(5)合成的影響Table 4 Effect of catalyst on the synthesis of compound (5)
2.1.4去甲烏藥堿-D4(6)的合成 使用氫溴酸作為脫甲基化試劑,產物色素較重,對產品的質控不利,而使用三溴化硼作為脫甲基化試劑有效避免了該問題。此外,在后處理過程中發現,因該化合物分子同時含有酸性基團(酚羥基)和堿性基團(氨基),體系的pH對結晶析出及收率至關重要,而且化合物含極性基團較多,其在水中具有一定的溶解性,水的使用量對其收率也有較大影響。
2.2 去甲烏藥堿-D4(6)的鑒定與表征
2.2.11H NMR 本文中所有化合物均通過1H NMR和HRMS進行鑒定,檢測結果與結構相符。去甲烏藥堿-D4(6)的1HNMR譜圖示于圖2。δ7.14 ppm,6.81 ppm兩個雙重峰為4-羥基苯甲基片段中苯環上的氫,6.63 ppm、6.62 ppm兩個單峰為1,2,3,4-四氫異喹啉結構中苯環上兩個氫,δ4.58 ppm峰為1,2,3,4-四氫異喹啉結構中1-位手性次甲基氫與相鄰的亞甲基耦合的核磁信號。與該手性中心相連的4-羥基苯甲基片段中芐位亞甲基上的兩個質子(圖3)為化學位移不等價,在δ3.37 ppm、2.97 ppm表現為兩個多重峰,與文獻報道一致[15]。去甲烏藥堿-D4(a)與非標記的去甲烏藥堿(b)的氫核磁譜圖對比示于圖4,由于四氫異喹啉3-位和4-位亞甲基上的質子被氘原子取代,因此在去甲烏藥堿-D4(6)的氫核磁譜圖上無信號峰。
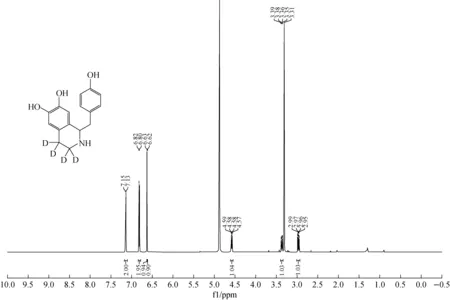
圖2 去甲烏藥堿-D4的1H NMR譜圖Fig.2 1H NMR spectra of higenamine-D4
圖3 模擬的去甲烏藥堿的立體結構Fig.3 Simulated 3D structure of higenamine
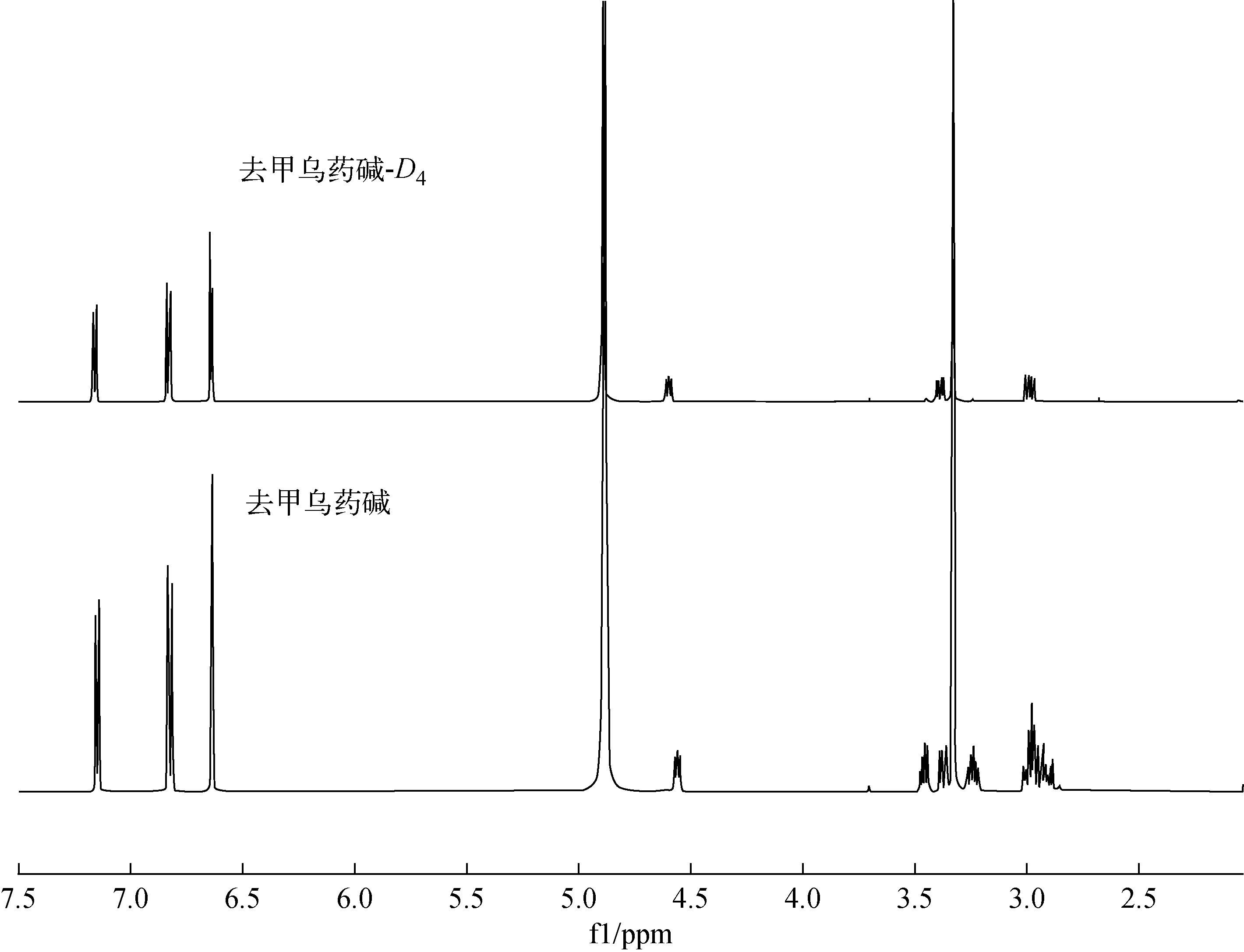
圖4 1H NMR核磁譜圖比對Fig.4 1H NMR comparison spectra of higenamine-D4 and higenamine
2.2.2HRMS確證及同位素豐度確認 去甲烏藥堿-D4的精確相對分子質量通過飛行時間質譜得到確認。通過質譜的結果(圖5)可知,目標化合物的分子離子峰[M+H]+=276.152 8,與理論計算結果[M+H]+=276.153 8相符。
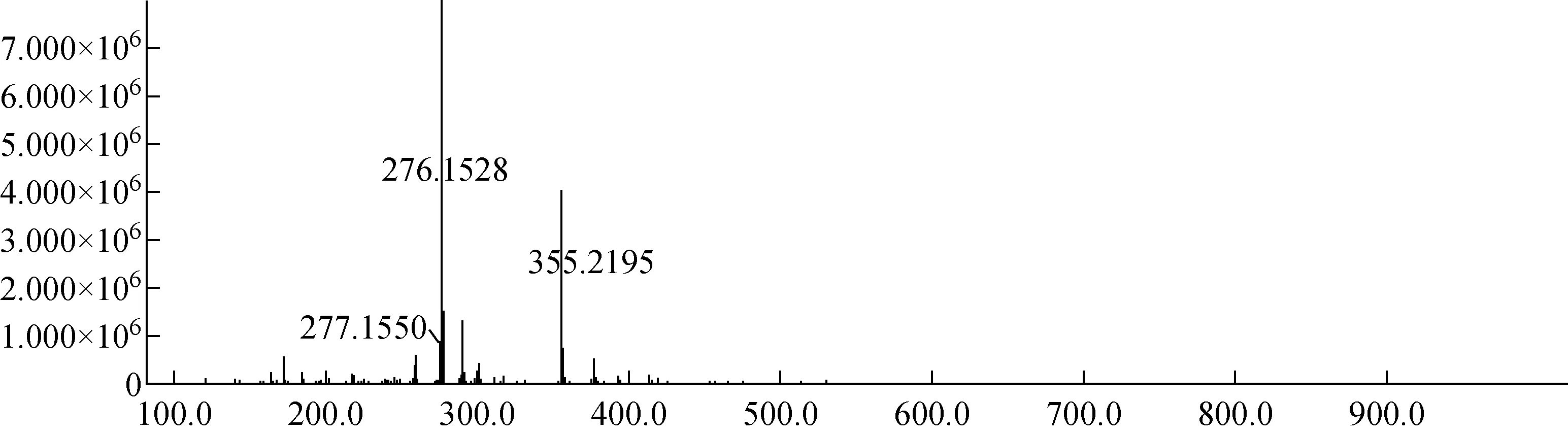
圖5 去甲烏藥堿-D4的HRMS譜圖Fig.5 HRMS spectrum of higenamine-D4
同時,產物經串聯質譜分析,用“質量簇分類法”[16-17]計算,去甲烏藥堿-D4氘同位素的豐度為96.5%,具體計算過程如下。
去甲烏藥堿-D4的質譜峰簇為:m/z=272~278,非氘標記的化合物天然豐度分布的占比(歸一化后)應為A272∶A273∶A274=0.84∶0.15∶0.01。將所采集到的質譜圖中m/z=272、273、274、275、276的峰強度數據歸一化后分別記為A0、A1、A2、A3、A4,代入方程組(1)計算,解xj(j=0、1、2、3、4)。
A0=0.84x0=0
A1=0.15x0+0.84x1=0
A2=0.01x0+0.15x1+0.84x2=0
(1)
A3=0.01x1+0.15x2+0.84x3=11.4
A4=0.01x2+0.15x3+0.84x4=72.6
從上述方程組中求得x0=0,x1=0,x2=0,x3=13.57,x4=84.01。
去甲烏藥堿-D4的同位素豐度值以E表示,按公式(2)計算得到同位素豐度,即氘原子標記率E=96.5%。
(2)
式中:xj表示標記j個D原子的去甲烏藥堿-Dj分子的摩爾分數,其中j=0、1、2、3、4。
3 結論
本文以2-(3,4-二甲氧基苯)乙腈為起始原料,以廉價易得的重水為穩定同位素標記源,經過氫-氘交換,再經氘代金屬還原劑還原得到關鍵中間體2-(3,4-二甲氧基苯)乙胺鹽酸鹽-D4,再經關環、脫保護基等步驟,合成了去甲烏藥堿-D4。其中關鍵是氫-氘交換和氘代還原兩個過程對化合物同位素豐度的控制,經過條件優化,最終合成得到的關鍵中間體的同位素豐度達到了預期,且后續反應并未有明顯的同位素豐度稀釋現象。本文所合成的目標化合物能作為內標試劑滿足去甲烏藥堿的定性與定量分析。
