腓骨肌萎縮癥一家系的臨床特征與突變基因分析
2021-08-24 02:51:10李爽朱梅佳唐吉友張小雨張霄劉小民
山東醫藥 2021年25期
關鍵詞:基因突變
李爽,朱梅佳,唐吉友,張小雨,張霄,劉小民
山東第一醫科大學第一附屬醫院(山東省千佛山醫院)神經內科,濟南250014
腓骨肌萎縮癥(CMT)是常見的遺傳性運動感覺周圍神經病之一,發病率約為1/2 500,其典型的臨床特征為肢體遠端進行性肌無力、肌萎縮、腱反射降低或消失以及感覺障礙等[1]。根據電生理和病理特征,CMT可分為兩型:CMT1型,又稱脫髓鞘型,其神經傳導速度減慢(正中神經運動傳導速度<38 m/s);CMT2型,又稱軸突型,其神經傳導速度正常或輕度減慢(正中神經運動傳導速度>38 m/s)。CMT具有高度的臨床變異性和遺傳異質性,目前已發現致病基因超過100個[2]。X-連鎖CMT1型(CMTX1型)是第二常見的CMT類型,僅次于CMT1A型,約占所有CMT類型的6.2%[3]。CMTX1型是編碼連接蛋白32(Cx32)的縫隙連接蛋白B1(GJB1)基因突變所致。2016年8月,山東省千佛山醫院收治1例CMT患者,現對該病例的臨床特征和突變基因進行回顧性分析。
1 臨床資料
先證者,男性,27歲。因“進行性四肢遠端肌無力伴肌萎縮1年”收入院。患者首發癥狀為行走困難,可獨立行走,但跑步能力下降,其后逐漸出現四肢遠端肌無力、肌萎縮,病情緩慢進行性加重,無肢體感覺異常。神經系統體格檢查:雙側前臂,雙手骨間肌、大小魚際肌,雙側脛前肌和腓腸肌以及雙足小肌肉呈不同程度萎縮,弓形足,雙上肢腱反射消失,雙下肢腱反射降低,無顱神經受累,無深淺感覺障礙,無共濟失調、肌張力障礙、錐體束征以及智能損害。腦脊液常規檢查:白細胞計數輕度升高(10×106/L)、總蛋白輕度升高(47.6 mg/dL),余未見異常。血生化檢查均未見明顯異常。神經電生理檢查:四肢運動和感覺神經傳導速度均減慢或引不出波,運動和感覺纖維均受累。肌電圖檢查:大量失神經電位和神經再生現象,呈神經源性損害。先證者神經電生理資料見表1、2。四肢周圍神經超聲、顱腦MRI未見異常。根據患者臨床表現和輔助檢查結果,懷疑為CMT[4]。
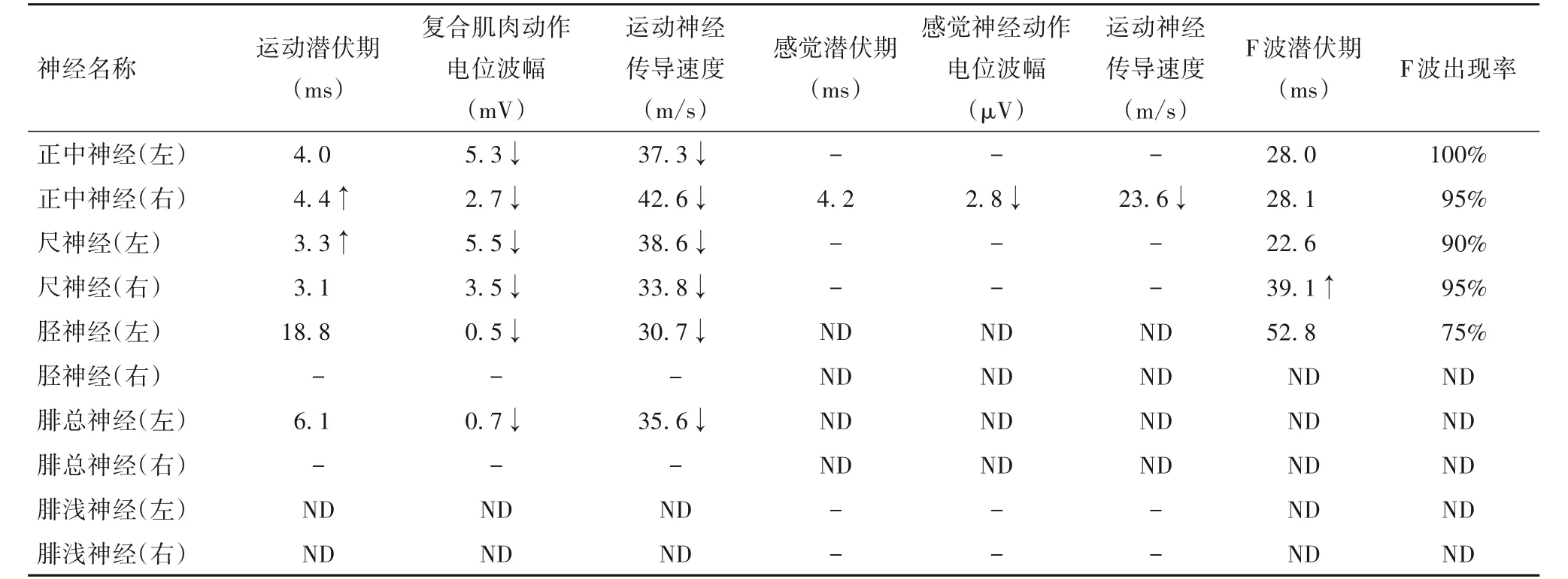
表1 CMT患者四肢運動和感覺神經電生理資料
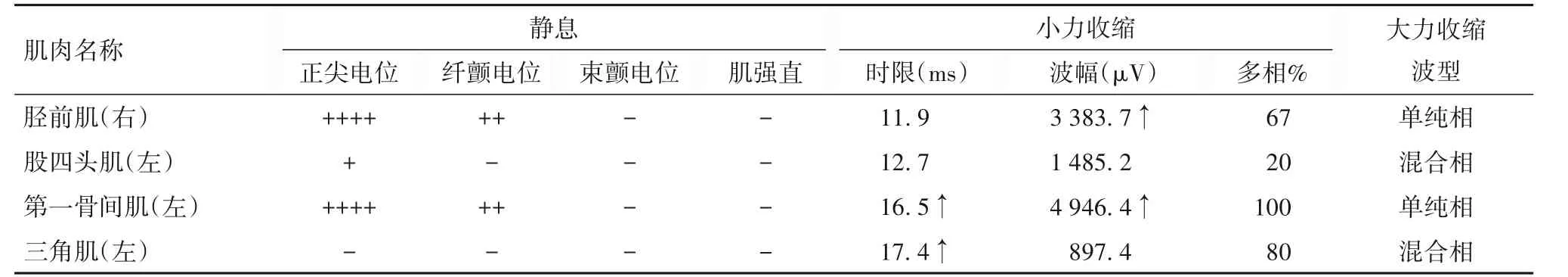
表2 CMT患者四肢肌肉肌電圖資料
經先證者及其家屬同意,對先證者的一級親屬進行家系調查。該家系共兩代4人,均為山東省常住居民,漢族,共發現1例CMT患者(即先證者)。先證者家系系譜圖見圖1。
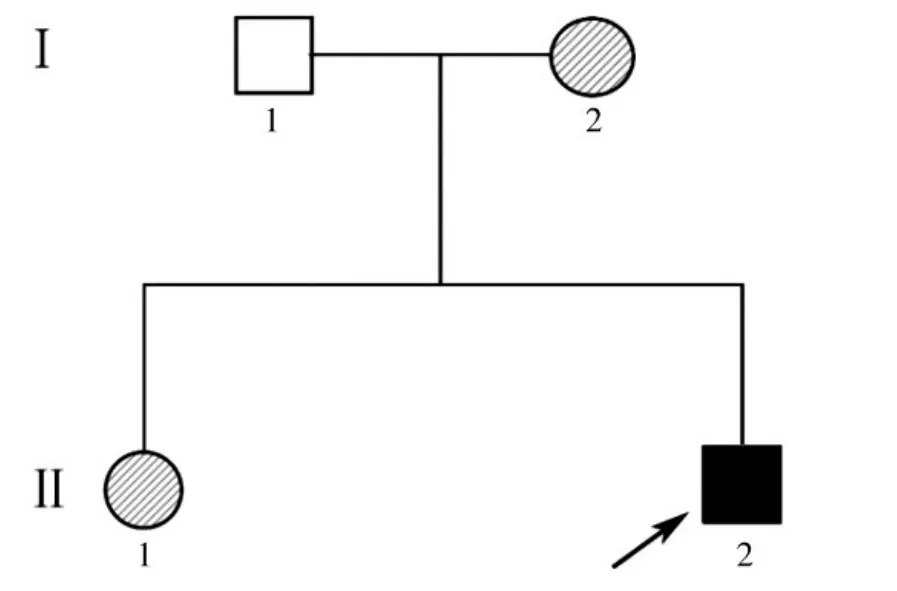
圖1 先證者家系系譜圖
為進一步明確診斷,采集先證者及其父母、姐姐外周靜脈血各2 mL,提取基因組DNA,采用多重連接依賴式探針擴增技術分析CMT突變基因,然后進行一代DNA測序驗證。結果發現,先證者同時存在GJB1基因第2外顯子c.44 G→A(Arg15Gln)半合子突變和外周軸蛋白(PRX)基因第7外顯子c.3208 C→G(Arg1070Gly)雜合變異;先證者姐姐同時存在GJB1基因c.44 G→A(Arg15Gln)雜合突變和PRX基因c.3208 C→G(Arg1070Gly)雜合變異;先證者母親存在GJB1基因c.44 G→A(Arg15Gln)雜合突變,PRX基因未檢測到變異;先證者父親存在PRX基因c.3208 C→G(Arg1070Gly)雜合變異,GJB1基因未檢測到變異。見圖2。
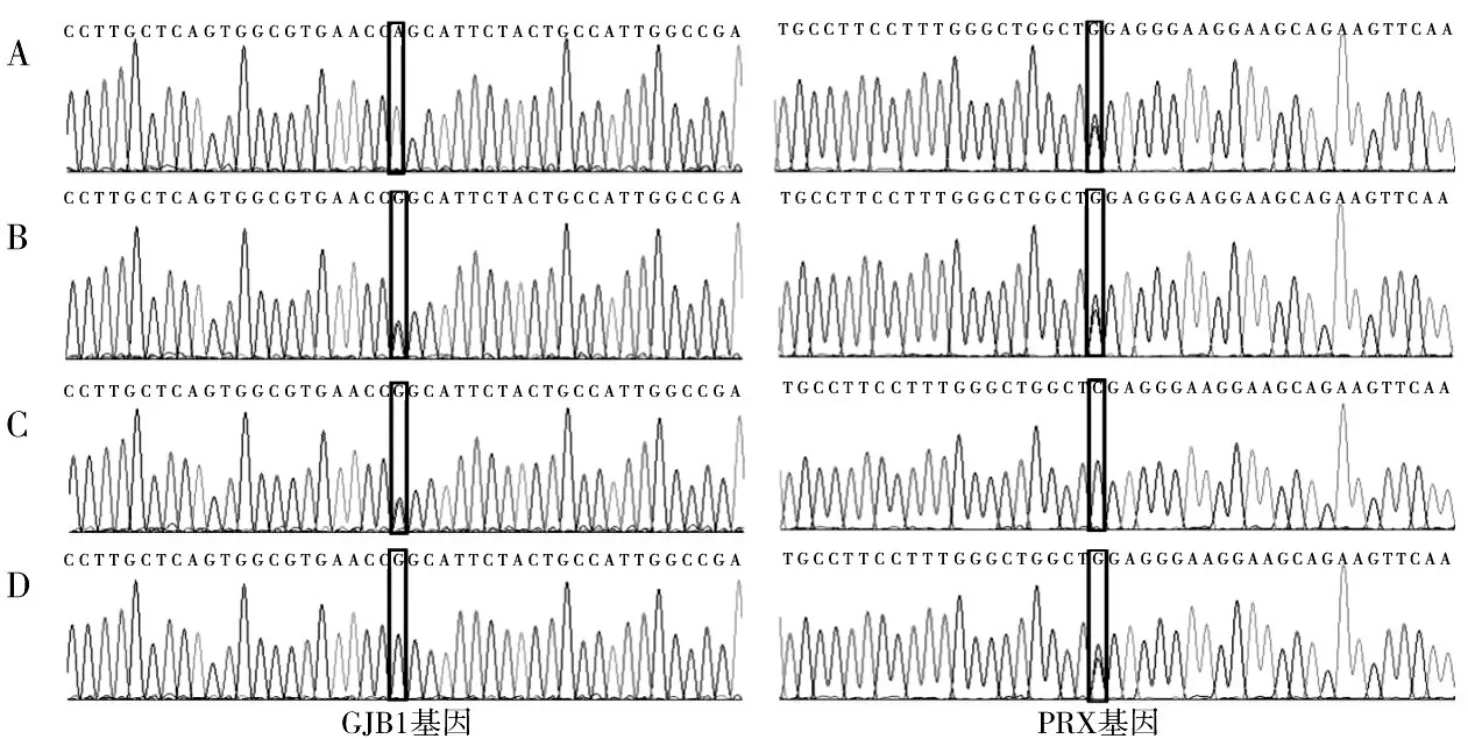
圖2 CMT家系各成員GJB1基因第2外顯子和PRX基因第7外顯子部分序列圖
2 討論
根據遺傳方式不同,CMT可分為常染色體顯性遺傳、常染色體隱性遺傳和X-連鎖顯性或隱性遺傳。本研究該家系內僅1例有臨床癥狀患者,根據家系調查結果難以確定遺傳方式,需要進行突變基因分析和DNA測序驗證。結果證實,該家系為GJB1基因c.44 G→A突變所致的顯性遺傳CMTX1型。CMT多為兒童或青少年發病,少數成年后發病。本例CMT患者發病年齡相對較晚,臨床表現主要為進行性四肢遠端肌無力伴肌萎縮,同時出現腱反射降低或消失,無深淺感覺障礙,但肌電圖檢查顯示四肢運動和感覺神經均已明顯受累。中間型CMT的正中神經運動傳導速度為25~45 m/s[5]。本例CMT患者左右正中神經運動傳導速度分別為37.3、42.6 m/s,介于CMT1型和CMT2型,符合中間型CMT表現。女性CMTX1型患者臨床癥狀輕重不一,甚至無癥狀,這種現象可能與女性髓鞘雪旺細胞的X染色體失活偏移有關[4,6]。在對先證者一級親屬的家系調查中發現,先證者姐姐和母親雖然攜帶GJB1基因c.44 G→A雜合突變,但無相關臨床癥狀,可能為亞臨床患者。TANIGUCHI等[7]觀察了30例CMT患者腦脊液蛋白水平,結果發現18例存在腦脊液蛋白水平升高,這可能與腦脊液循環障礙和神經根處血—神經屏障受損有關。本例CMT患者腦脊液檢查發現,白細胞計數和總蛋白水平輕度升高,亦提示存在周圍神經損害。除典型的臨床表現外,少數CMTX1型尚可出現中樞神經系統受累,以男性多見,主要表現為偏癱、構音障礙、吞咽困難、共濟失調、精神癥狀、意識障礙等,癥狀持續時間不一,多在數小時至數周消失;顱腦MRI檢查可見異常融合的T2、FLAIR、DWI高信號,常見于腦白質和胼胝體,這些異常信號消失時間較長,但多數為可逆性白質改變,這可能是由于突變的Cx32導致腦白質可逆性軸突損傷引起的[4,6,8]。本例CMT患者無中樞神經系統受累癥狀,顱腦MRI檢查無異常,提示無中樞神經系統受累。
在我國CMT患者中,由GJB1基因突變所致的CMTX1型占7.32%。目前,已發現超過450個GJB1基因突變,大多數是錯義突變,其余為缺失或移碼突變。近期LIU等[8]對中國465個無親緣關系的CMT患者進行了GJB1基因突變分析,結果發現24個錯義突變、4個無義突變、1個缺失突變、1個內含子突變和4個框移突變。本研究基因突變分析發現,該家系先證者及其姐姐同時存在GJB1基因c.44 G→A半合子突變和PRX基因c.3208 C→G雜合變異,前者來源于其母親,后者來源于其父親,其中GJB1基因c.44 G→A半合子突變為該家系的致病性突變。PRX基因純合突變可導致更為少見的常染色體隱性遺傳CMT4F型[9],本研究發現的PRX基因c.3208 C→G為雜合變異,考慮為非致病性,該雜合變異國內外鮮見報道,千人基因組、HGMD、ESP6500、dbSNP等數據庫亦未收錄,可能為一新的PRX基因變異。
GJB1基因定位于人染色體Xq13.1,編碼Cx32。生理狀態下,Cx32在周圍神經髓鞘內高表達,它在髓鞘內形成縫隙連接,作為神經營養物質、細胞因子以及各種離子和細胞信號轉導分子的轉運通道,對髓鞘的形成和維持起到至關重要的作用。突變的Cx32蛋白喪失了縫隙連接和物質轉運通道的功能,可導致周圍神經脫髓鞘和軸突變性[1,8,10]。PRX基因定位于人染色體19q13.13-13.2,編碼Periaxin蛋白。生理狀態下,Periaxin蛋白通過與肌營養不良相關蛋白2和肌營養不良蛋白形成復合體,對維持周圍神經髓鞘的穩定性具有重要作用。該基因突變后,髓鞘穩定性受到破壞,并且導致髓鞘再生障礙,最終引起周圍神經嚴重脫髓鞘而發病[11]。該基因突變后可導致常染色體隱性遺傳CMT4F型[9]。CMT4F型多為嬰幼兒發病,臨床特征主要為運動發育遲緩、肌無力、肌萎縮、感覺障礙、感覺共濟失調、腱反射降低或消失,電生理檢查提示神經傳導速度<10 cm/s或測不出[9,12]。本研究在同一家系同一患者中同時存在上述兩個基因突變,但先證者臨床表現與CMT4F型不符,無法確定PRX基因突變是否參與CMTX1型發病。
綜上所述,本例CMT患者臨床特征主要為緩慢進行性四肢遠端肌無力伴肌萎縮,同時出現腱反射降低或消失;腦脊液檢查白細胞計數和總蛋白水平輕度升高,肌電圖檢查四肢感覺和運動傳導速度減慢或引不出波;本例CMT患者同時存在GJB1基因c.44 G→A半合子突變和PRX基因c.3208 C→G雜合變異,GJB1基因c.44 G→A半合子突變為其致病性突變。
猜你喜歡
英語世界(2023年6期)2023-06-30 06:29:10
中國醫學影像學雜志(2021年6期)2021-08-13 08:43:36
中國生殖健康(2020年2期)2021-01-18 02:51:26
小學生導刊(2018年13期)2018-06-29 03:49:00
中國生殖健康(2018年2期)2018-01-12 13:57:51
現代檢驗醫學雜志(2016年4期)2016-11-15 02:01:14
中國現代醫學雜志(2015年26期)2015-12-23 11:04:22
鄭州大學學報(醫學版)(2015年2期)2015-02-27 14:50:44
中華皮膚科雜志(2014年4期)2014-12-19 12:55:49
中國神經精神疾病雜志(2014年1期)2014-03-01 03:23:22
