載鈀硅藻土恒流吸氫特性
2021-08-26 02:58:08于廣琛王曉冬
中國新技術新產品 2021年10期
關鍵詞:實驗
于廣琛 王曉冬
(東北大學,遼寧 沈陽 110000)
氫能源在國防和工業領域起著舉足輕重的作用。在氫同位素分離技術中,鈀是運用最多的分離材料[1]。鈀優先吸附較輕的氫同位素而不是較重的氫同位素,該效應在低溫下比在高溫下更明顯。且其不易被環境氣體鈍化,抗中毒能力強,利用氫氣很容易在較低的溫度下使其活化,具有很好的吸放氫動力學特性,因此將鈀作為氫同位素分離的首選材料。在分離工藝中,因為要求在低溫條件下的一定時間內以一定流量進氣,所以鈀-氫體系的恒流動力學性質對美國新型TCAP工藝起著決定性的作用。對于Pd-H體系來說,國內外已有的相關研究主要集中在鈀的熱力學、鈀納米顆粒的制備和改性以及輻照對鈀吸放氫性能的影響等方面[2]。對于鈀吸氫動力學特性的研究較少,而對于將Pd附著在硅藻土上進行恒流吸氫動力學的研究尚未見文獻報道。該文旨在探究將載鈀硅藻土(Pd/K)作為TCAP分離柱材料,對在TCAP工藝溫度下進行恒流吸氫的動力學進行研究,為TCAP工藝及載鈀硅藻土-氫體系的工程提供理論和一定的支撐數據。
1 實驗部分
1.1 實驗系統
為滿足實驗要求,自制一套Pa量級的高精度測量的真空系統(如圖1所示),載鈀硅藻土在273 K下的平衡壓很低(在1 kPa以下),測量樣品室端的真空測量裝置采用0 kPa~1 kPa的電容膜片真空計,儲氣罐端采用0 kPa~100 kPa的電容膜片真空計,精度均為0.003%。實驗用量程為1 sccm質量流量控制器。低溫設備采用低溫恒溫槽,溫度范圍為-60 ℃~100 ℃,溫度浮動±0.05 ℃。氫源由LaNi4.25Al0.75供氣床提供。為使反應器體積盡可能小,需要減少材料堆積,采用自制304不銹鋼薄壁錐型反應器。整個系統可達10-5Pa量級真空度。

圖1 恒流吸氫裝置
1.2 實驗樣品
Pd/K材料由北京有色金屬研究總院提供,合成的Pd/K材料依然保持原硅藻土顆粒不規則的球形形貌。化學成分為Pd、SiO2,其中Pd質量分數為56%;顆粒形貌為50目~60目;Pd/K經過1200 ℃ 熱處理1 h;為避免鈀吸氫放熱對實驗結果造成影響,樣品裝量約1 g。
實驗前需要對樣品進行活化處理,將Pd/K樣品裝入反應器,300 ℃抽真空除氣2 h,降至室溫飽和吸氫;再次加熱至200 ℃放氫,充分放氫后,降至室溫繼續吸氫;重復吸、放氫3~5次,活化完畢。
1.3 實驗方法
實驗方法如下:1) 將流量計調至全開,系統抽真空至1.0×10-4Pa。2) 停止抽真空,關閉流量計及其前后的閥門,將反應器放入低溫恒溫槽,待反應器冷卻。3) 打開儲氣罐開關引入氫氣,再調節流量控制器向反應器通入氫氣,通過計算機自動采集實時壓力數據。
1.4 恒流法動力學方程模擬
根據得到的恒流等溫吸放氫動力學曲線,采用Chou模型、JMAK模型或Jander模型對吸放氫動力學進行擬合嘗試。由于α相區吸氫量過少,β相壓力高(超過真空計量程),因此只對α+β相區進行擬合,并考慮壓力的變化情況。
2 實驗結果與討論
2.1 恒流吸氫PCT曲線
273 K以下Pd/K吸氫坪臺壓低于1 kPa,流量計下游體積約12 mL,因此實驗中認為進氣量即吸氫量,據此繪制恒流吸氫PCT曲線。實驗對Pd/K材料在233 K、243 K、253 K以及263 K條件下進行流量為0.3 sccm/g、0.5 sccm/g、0.7 sccm/g、1.0 sccm/g以及1.5 sccm/g的小流量恒流量測試。
以233 K為例,如圖2所示,當溫度恒定時,Pd/K吸氫速率增加而坪臺區的斜率基本保持不變。坪臺壓力隨流量增大逐漸升高。當進氣速率增大時,吸氫速率增大,這就要求外界有更高的壓力作為驅動力促使Pd/K吸收氫氣,因此宏觀表現為壓力升高。

圖2 流量對動態平衡壓的影響
以0.5 sccmg-1為例,由圖3可知,隨著溫度增加,坪臺區斜率也逐漸增加。這是由于Pd/K吸氫為可逆反應,進氣速率(吸氫速率)由質量流量控制器控制并保持恒定,而逆反應受溫度及壓強的控制,坪臺區壓強較低,當溫度增加時氣體釋放速率也隨之增加,此時凈吸氫速率小于進氣速率,壓力逐漸上升。因為溫度恒定,壓強又較低,即使不同流量條件下逆反應速率也基本一致;所以不同進氣流量下Pd/K坪臺區壓力曲線的斜率基本一致。由α+β相向β相轉變的區域逐漸變大。隨著溫度增加,由α+β相向β相的轉變區域由尖銳變得越來越圓滑。隨著溫度的增加,坪臺區壓力逐漸增大,這表明,隨著溫度和氣相壓力的增加或者是由于內應力的作用,金屬內部仍處于固溶體的氫原子的化學勢增大,使由固溶體相向氫化物相的轉變變得更加困難,在曲線上表現為相對于溫度低時,溫度高時的過渡區域的出現更加提前,壓力也提前上升,這就使過渡區曲線變得更為圓滑。

圖3 溫度對動態平衡壓的影響
2.2 動力學曲線
2.2.1 平衡壓方程
該文采取多項式擬合結合Van`t Hoff方程對Pd/K的吸氫平衡壓進行預報。該方法的通用方程如公式(1)所示。

式中:Rg為氣體狀態常數,Tref為進行平衡壓多項式擬合時采用的參考溫度;f(H/M)為參考溫度下PCT曲線的多項式擬合方程,代表參考溫度下平衡壓與H/M的關系;Peq為平衡壓,代表在PCT曲線內某一氫金屬比下吸氫或放氫達到平衡時的壓力。
平衡壓方程由參考溫度下的PCT曲線在Origin內進行11階多項式擬合得到,如公式(2)所示。

式中:aiH/M i為11階多項式擬合項;Peq,ref為參考溫度下的平衡壓。
對于Pd/K的熱力學性質,根據吸氫PCT實驗數據,選擇263 K下的實驗點作為參考溫度下的Peq-H/M關系f(H/M),對該溫度下的實驗點進行多項式擬合。其中f(H/M)的單位為Pa。已知其吸氫焓變值ΔH為-33.71 kJ·mol-1H2。擬合平衡壓方程曲線如圖4所示。
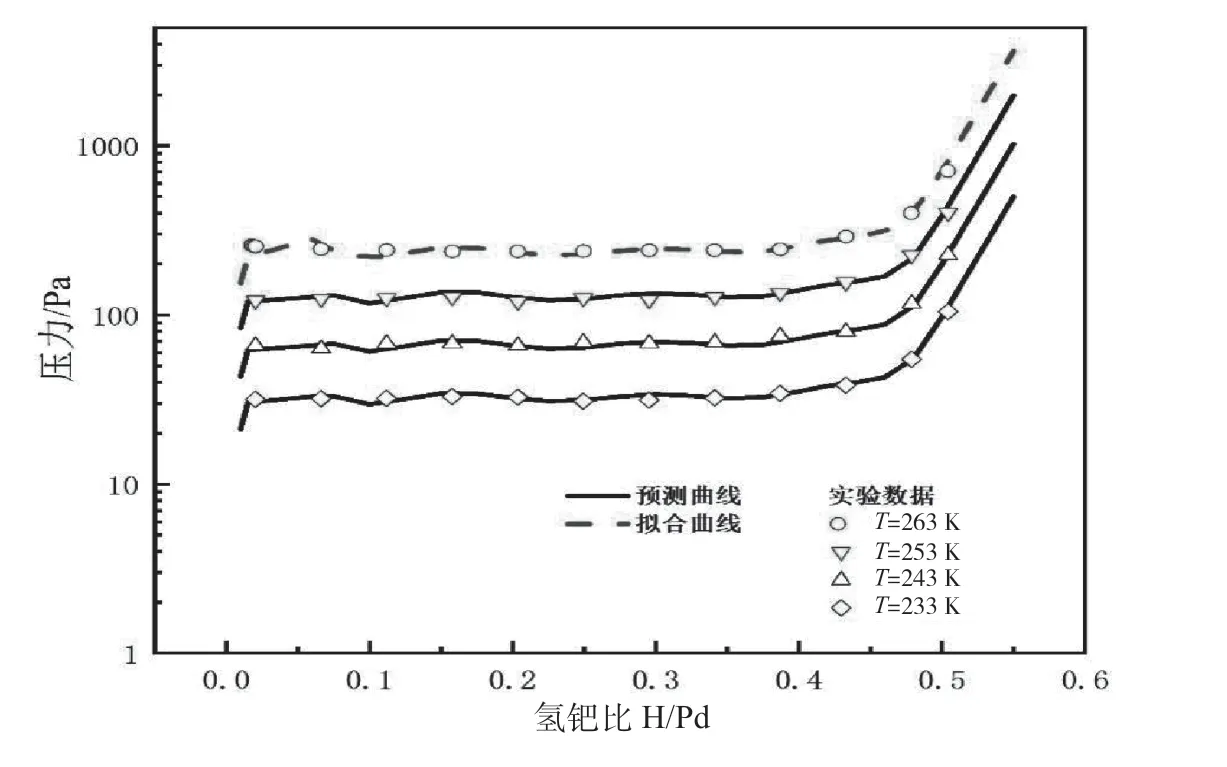
圖4 平衡壓方程曲線
2.2 動力學曲線分析及預測
在233 K~263 K對0.3 sccm/g~1.5 sccm/g流量范圍內選取動力學方程進行擬合[3]。發現與(t為時間)均呈線性關系,這表示表面滲透在該階段為限制反應速率的控速步驟。
根據擬合得到的速率常數Kf,建立Kf與流量F的關系,在253 K條件下,在0.3 sccm/g~1.5 sccm/g流量范圍內,Kf與流量F呈線性關系。以253 K為例下Kf與F的關系如公式(3)所示。

Kf不僅受流量F的影響,同時也受溫度T的影響。Kf隨溫度增大而明顯增大。因此,Kf的關系如公式(4)所示。
式中:A為常數;Tref為參考溫度;f(F)為Kf與F的函數。
分別在0.3 sccm/g、0.5 sccm/g、0.7 sccm/g、1 sccm/g以及1.5 sccm/g對lnKf與1/T進行線性擬合。擬合得斜率分別為-2034.8、-2132.0、-2203.0、-2169.0、-2062.0,其平均值為-2120.0。根據阿倫尼烏茲方程,斜率乘以Rg即為Pd/K坪臺區表觀的活化能Ea=-17.63 kJ·mol-1。綜上得到系數Kf與流量F,溫度T的關系式,如公式(5)所示。

預測在0.3 sccm/g~1.0 sccm/g時,各個溫度準確性都較高,預測曲線最大偏差在40 Pa以內。氫金屬比為0.025~0.050的預測曲線出現向下波動的情況,這與擬合多項式方程有關。根據預測得到的Kf值,以263 K為例,預測Pd/K吸氫坪臺壓力力曲線,如圖5所示。

圖5 263 KPd/K吸氫PCT實驗及預測曲線
根據實驗及擬合結果可以判斷,Pd/K在恒流0.3 sccm/g~1 sccm/g及233 K~263 K條件下,吸氫平臺區受表面滲透控速步驟控制,在溫度和流量逐漸增加的條件下,控速步驟并未發生改變。低溫Pd/K吸氫坪臺壓力較低,氫分子在表面吸附解離放熱,并在濃度差作用下滲透進入鈀晶格內,這表明Pd/K材料的動力學性能較好,形核長大過程并未限制其反應速率。
對于Pd/K恒流吸氫實驗,質量流量控制器下游體積在20 mL以下,吸氫坪臺區壓力較低(1 kPa以內),該體積內的氫氣摩爾量可以忽略,因此進氣量即為吸氫量,氫金屬比與時間呈線性關系,如公式(6)所示。

式中:M為Pd的相對原子質量;m為Pd/K質量;ω為Pd含量;Vm為標準摩爾體積;F為流量;t為時間。
因此,在坪臺區壓力P與氫金屬比H/M與流量F及溫度T的關系如公式(7)所示。

3 結論
對恒流吸氫實驗數據分析擬合,得到以下結論:1) Pd/K在233 K~263 K、流量為0 sccmg-1~1.5 sccmg-1的條件下,吸氫控速步驟為表面滲透,并得到其坪臺區表觀活化能為-17 kJ·mol-1H2。2) 獲得了恒流條件下Pd/K吸氫壓力與氫金屬比、溫度以及流量的關系式,并進行了預測,與實驗數據接近,預測準確。3) 測得的數據及分析結果對TCAP工藝尤其是在壓力、流量的選取工作中發揮了重要作用。
猜你喜歡
作文·小學低年級(2025年2期)2025-02-13 00:00:00
小雪花·小學生快樂作文(2024年11期)2024-12-31 00:00:00
作文·小學低年級(2024年2期)2024-04-29 00:00:00
作文·小學低年級(2023年3期)2023-04-29 00:00:00
小獼猴智力畫刊(2022年9期)2022-11-04 02:31:42
小主人報(2022年4期)2022-08-09 08:52:06
中學生數理化·中考版(2022年11期)2022-02-16 07:01:20
小哥白尼(趣味科學)(2019年6期)2019-10-10 01:01:50
發明與創新(2016年38期)2016-08-22 03:02:52
太空探索(2016年5期)2016-07-12 15:17:55
