助催化劑強化電芬頓技術去除水中難降解有機物的研究進展
2021-09-07 08:43:10鄭豪程松沈晨于偉華劉福強
土木與環境工程學報 2021年6期
關鍵詞:催化劑
鄭豪,程松,沈晨,于偉華,劉福強,
(1. 南京大學 環境學院;污染控制與資源化研究國家重點實驗室,南京 210023;2. 南京環保產業創新中心有限公司,南京 211106)
化工、醫藥、農藥等行業生產過程中產生大量含多環芳烴、鹵代烴、雜環類化合物、農藥、抗生素等難降解有機物的工業廢水,通常具有高毒性、高鹽分等特征,已成為工業水處理領域的難點[1-2]。
對于工業廢水中難降解有機物,通常采用氧化法、物化法等技術處理。近年來,高級氧化技術因作用效率高、適用范圍廣等優勢而獲得快速發展,其中以羥基自由基(HO·)為主導的芬頓氧化技術受到持續關注,但因H2O2利用率低、Fe2+用量大以及大量鐵泥后續處置難等問題,限制了其進一步應用推廣[3]。
電芬頓技術將電化學與芬頓技術相結合,主要優勢包括:可原位生成H2O2,降低H2O2在運輸、存儲等過程的安全風險,緩解了一次性投加H2O2導致的自分解問題;陰極提供電子,將Fe3+有效還原為Fe2+,大幅減少鐵泥產量;實現陽極氧化、電吸附等協同作用,顯著提高有機物去除效率[4-6]。
筆者基于電芬頓原理、局限性以及強化方法,著重分析助催化劑強化電芬頓技術的原理、特性并展望了改良方向。
1 電芬頓及其強化方法
1.1 電芬頓原理
電芬頓技術以Fe2+和電化學產生的H2O2作為芬頓試劑的來源[7],通過Fe2+與H2O2發生芬頓反應生成HO·,從而實現污染物的高效降解,如式(1)~式(5)所示。

(1)

(2)
(3)
Intermediate products
(4)
CO2+H2O+Inorganic ions
(5)
電芬頓可以分為均相電芬頓和非均相電芬頓兩大類。其中,均相電芬頓中Fe2+與H2O2在溶液中發生均相反應產生活性氧物種,而非均相電芬頓則以鐵活性物種或將其負載至載體作為鐵源催化劑,在其表面發生非均相反應并產生自由基。
電芬頓去除難降解有機物的效率主要受3方面因素限制:1)pH值適用范圍窄,通常需要酸性條件(pH≈3.0);2)陰極H2O2產率和電流效率低,限制了芬頓反應速率;3)Fe3+/Fe2+的循環速率是生成羥基自由基降解污染物的主要限速步驟。
1.2 電芬頓強化方法
為提高電芬頓技術對難降解有機物的去除效率,研究和應用較多的強化方法主要包括3方面:1)優化設計反應器,通過增強傳質使污染物與活性氧充分接觸[4];2)載體固定催化劑,能夠提高催化劑的穩定性、減少金屬離子的浸出,同時,催化劑分散在載體上,活性位點增加,可以提高催化活性;3)添加助催化劑,包括直接投加到電解液中或同催化劑一起制備到陰極上,能有效增加Fe2+的含量或提高H2O2的選擇性生成,提高催化劑的活性、穩定性等。
近年來大量研究發現,添加少量助催化劑即可有效提升電芬頓對難降解有機物的去除效果[8-11]。研究較多的3種助催化手段及其機理如表1所示。

表1 電芬頓助催化手段及機理Table 1 Co-catalysis methods and mechanisms of EF
2 助催化劑強化電芬頓技術
電芬頓涉及的催化反應主要為陰極催化O2通過兩電子氧還原反應(2e-ORR)生成H2O2以及Fe2+催化H2O2生成HO·。為增強催化劑的催化作用,可通過優選優用助催化劑,直接或間接增強電芬頓技術性能。
2.1 改善鐵離子穩定性
鐵在較高pH值條件下會沉淀形成大量鐵泥,導致有效鐵含量減少,進而削弱污染物的去除效率。鐵離子與配體的配位作用可以保證其在接近中性pH的條件下以離子形態存在,因此,添加配體能拓寬有效pH范圍。而且,絡合物可通過一系列反應產生更多自由基,從而增強電芬頓去除有機污染物的性能。其反應機理如圖1所示。
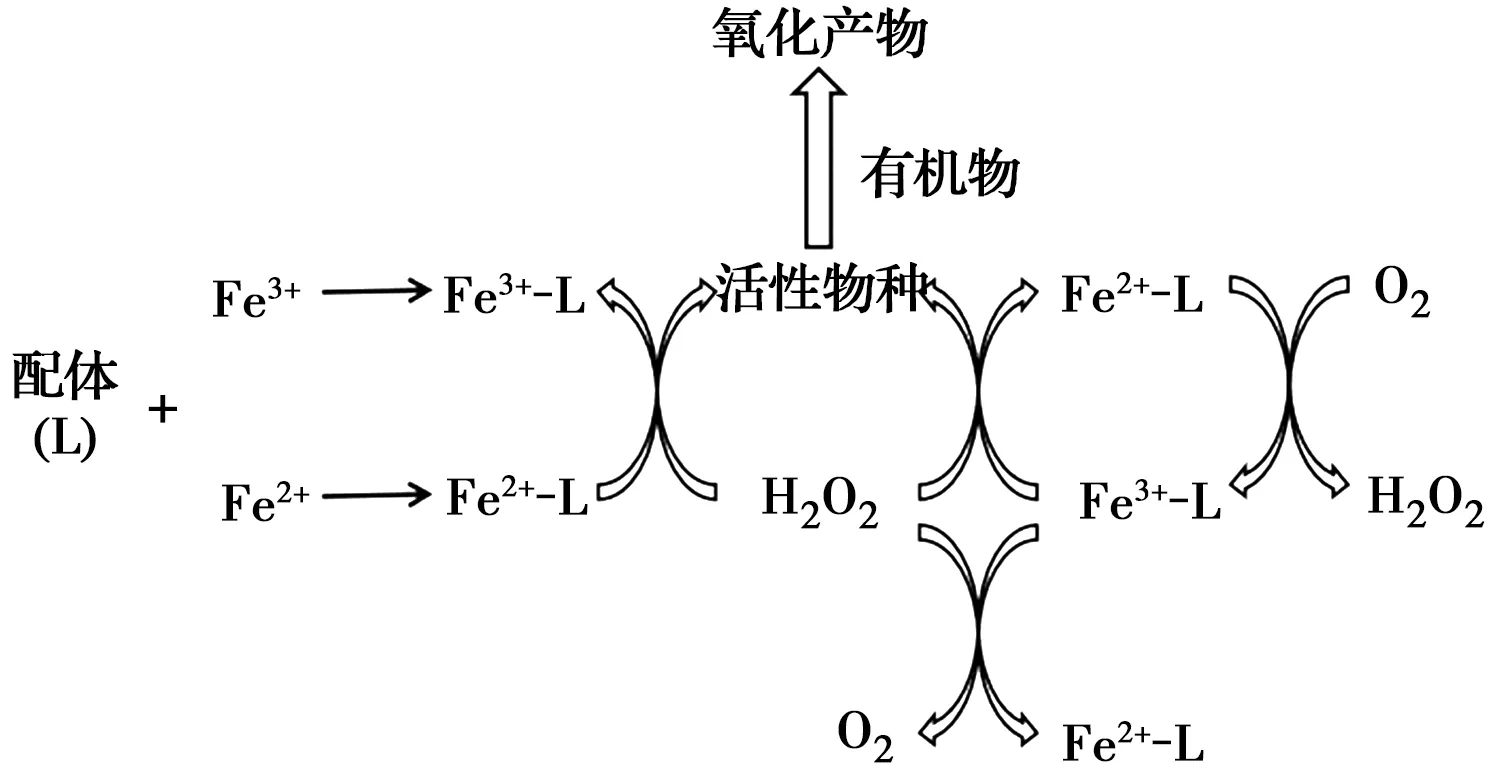
圖1 配體強化芬頓反應機理Fig.1 Mechanism diagram of ligand enhanced
乙二胺四乙酸(EDTA)、氮三乙酸(NTA)、乙二胺二琥珀酸(EDDS)、羥胺等有機配體被廣泛應用到芬頓體系中,可在pH>3.0的條件下有效去除難降解有機物。Zhang等[12]將NTA加入電芬頓體系,pH=5.0~8.0下均可在20 min內完全去除水中苯酚,速率常數約為0.26 min-1,較不加助催化劑提高了近63%。強絡合劑EDDS是一種具有生物降解性的有機配體,與NTA相比,對環境的二次污染更小,Ye等[13]將其引入電芬頓體系,以Fe(Ⅲ)-EDDS為可溶性催化劑,pH=9.0下僅約6%的鐵發生沉淀,而未添加EDDS時89%以上的鐵發生沉淀。不同的Fe-L(配體)絡合物的氧化還原性能存在差異,Liu等[14]結合理論計算發現,[Fe(Ⅲ)-EDTA]-絡合物的電子還原速率約是[Fe(Ⅲ)-EDDS]-絡合物的兩倍,更強的得電子能力有助于加速鐵循環,進而促進電芬頓性能。除了拓寬pH值范圍外,部分有機配體不僅作為螯合劑,同時作為還原劑,利用配體-金屬間的電荷轉移,促進Fe3+還原為Fe2+,從而增強電芬頓效率[15-17],Hou等[18]發現在芬頓體系中添加羥胺后HO·的生成速率常數是未添加時的100~10 000倍。然而,有機配體投加會增加有機負荷,進一步增加處理成本。此外,在處理過程中產生的活性物質會氧化分解有機配體,從而降低活性及穩定性[19]。
為此,近年來無機配體頗受重視。Wang等[20]用四聚磷酸鈉(Na6TPP)作為電芬頓體系電解質,形成的鐵-四聚磷酸鹽絡合物(Fe-TPP)保證鐵以離子形態存在于溶液中,可以在pH值4.0~10.2的范圍內有效降解阿特拉津(ATZ)。聚磷酸鹽雖然不會消耗HO·,但其使用會導致后續除磷難題。Cui等[21]采用環境友好的無機配體二硅酸鈉(SD)增強鐵電解系統,Fe2+/Fe3+-SD絡合物的生成有效避免了鐵離子的水解沉淀,因此,可在pH值為5.0~8.0條件下穩定運行。另一方面,鐵離子與SD絡合,可以有效降低Fe3+/Fe2+的氧化還原電位,使O2的還原熱力學過程更有利[22]。
綜上,多種有機和無機配體均可有效拓寬電芬頓體系pH適應范圍,并在近中性條件下獲得優良去除效果,如表2所示。然而,鐵配合物的重復利用以及后續處理問題亟待解決。Jin等[23]發現用載體固定鐵絡合物可提高重復利用率,通過將鐵-二吡啶甲酰胺絡合物(Fedpa)固定在SiO2上成功制備Fedpa@SiO2催化劑,重復利用3次后,仍能去除90%以上的2,4-二氯苯酚(2,4-DCP)。
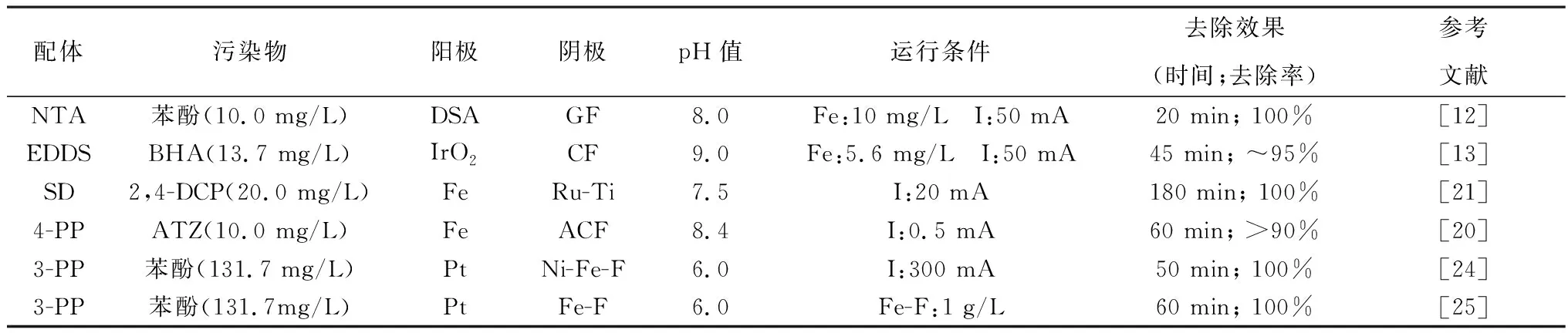
表2 配體強化電芬頓體系去除難降解有機物的效果Table 2 Effect of ligand enhances EF system in removing refractory organics
目前,對于配體強化電芬頓技術的研究仍集中在單一配體的作用機制。實際廢水水質復雜,通常同時存在多種有機配體和無機配體,闡明不同配體之間的相互作用規律與機制,可為直接利用水體中存在的配體提供重要參考,進而減少外源投加,降低可能造成的二次污染風險。
2.2 提升Fe3+/Fe2+循環速率
Fe3+還原為Fe2+的速率直接影響了電芬頓中HO·的生成速率以及污染物的降解速率,因此,加速Fe3+/Fe2+循環一直是研究熱點。添加能夠改善催化劑電子傳遞性能的助催化劑,可以加速Fe2+的再生。目前研究較多的是雙金屬協同催化技術。
在Fe-M(M為其他金屬)雙金屬體系中,利用雙金屬協同效應可以促進Fe2+再生,機理圖如圖2所示。金屬硫化物是研究較多的助催化劑,其表面不飽和的S原子捕獲質子形成H2S后,暴露具有還原性的金屬活性位點,從而加速Fe3+/Fe2+循環,Li等[26]在電芬頓體系中投加WS2后,羅丹明B(RhB)降解的速率常數提升了63.1%,這得益于W4+快速還原Fe3+。Mo是工業上常用的低毒性催化劑,Tian等[27]用MoS2助催化電芬頓技術去除磺胺甲嘧啶(SMT),污染物去除的表觀速率常數從原先的0.26 min-1增加到0.54 min-1。往反應池中直接投加助催化劑存在難以重復利用、增加運行成本的問題,因此,將過渡金屬與Fe一起制備成復合材料得到了學者們的關注。
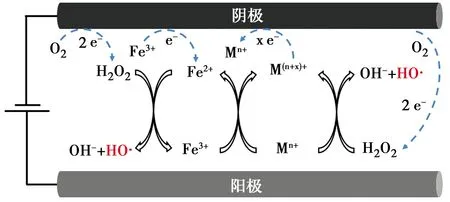
圖2 Fe-M雙金屬催化劑電芬頓體系機理Fig.2 Mechanism diagram of electro-Fenton EF system with
Cu是一種導電性好、廉價的過渡金屬,因而應用廣泛,多種鐵銅雙金屬催化劑被成功用于電芬頓體系,如表3所示。Luo等[28]通過兩步還原法制備了Cu摻雜的Fe@Fe2O3(CFF)納米粒子,Cu/Fe質量比為50%時,2 h內四環素(TC)去除率較nZVI提升了近12%。Barros等[29]通過共沉淀法合成Fe3-xCuxO4(0≤x≤0.25)NPs,由于鐵和銅離子在尖晶石結構八面體表面位置的協同作用,Cu2+/Cu+表面物種的存在對紫紅花食用染料的降解有顯著促進作用,Fe2.75Cu0.25O4體系的TOC去除率遠高于Fe3O4體系,前者約為70%,而后者僅不足39%。由于Cu本身也能催化H2O2生成HO·,因此,鐵銅協同作用的相對貢獻需要進一步計算。Ren等[30]利用一種新亞銅試劑捕獲Cu(Ⅰ),屏蔽Cu(Ⅰ)與鐵和H2O2的相互作用,從而定量鑒別各因素對增強芬頓反應催化活性的貢獻,其中19%的增強作用被確定為鐵銅協同作用。Co是一種具有類芬頓活性的過渡金屬,Ganiyu等[31]在碳氈(CF)上生長了分級CoFe-層狀雙氫氧化物(CoFe-LDH)并制成陰極,可在較寬的pH值范圍內(pH值為2.0~7.1)實現酸性橙II (AO7)的有效礦化,在pH值為3.0的條件下2 h內TOC去除率可達87%以上,Co2+的協同催化作用促進了Fe2+再生和HO·生成。
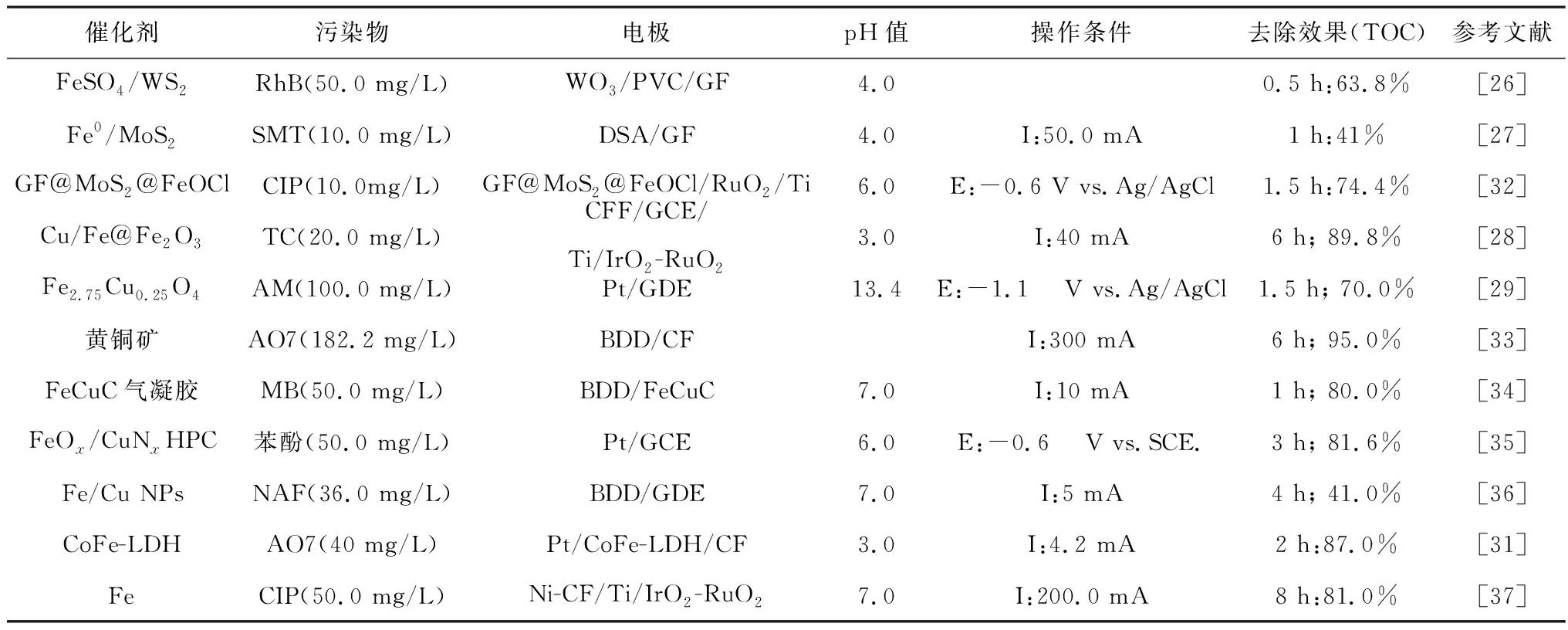
表3 Fe-M雙金屬催化劑電芬頓體系去除難降解有機物的效果Table 3 Effect of EF system with Fe-M bimetal catalyst in removing refractory organics
除了直接促進Fe3+還原,部分金屬助催化劑通過生成其他物質來促進Fe3+/Fe2+循環。Liu等[37]制備了Ni負載CF陰極(Ni-CF)并用于強化電芬頓降解環丙沙星(CIP),Ni的引入能夠明顯改善TOC去除率,8 h的TOC去除率可從約42%(CF)提升至81%(Ni-CF)以上,這主要歸因于Ni涂層誘導產生的大量還原性氫原子(H*)能提供電子能有效還原三價鐵,促進鐵循環過程,如式(5)所示。

(5)
綜上,雙金屬協同效應對電芬頓效率的提升十分顯著,但金屬離子浸出以及可能造成的二次污染仍不可忽視,將過渡金屬固定在載體上(常用的有碳材料)的復合催化劑能提高金屬的穩定性,然而金屬的負載會顯著影響H2O2的生成。Cheng等[38]制備了一種OCNT封裝Fe-Co-85 PBA的鎧甲式催化劑,能夠保證H2O2高效生成與活化,同時,碳層的保護作用進一步減少了金屬離子的泄露。未來雙(多)金屬催化劑的研究重心仍在于研發合適的載體及其有效的復合方式,使其兼具芬頓活性位點以及兩電子氧還原活性位點。此外,納米形式的雙金屬催化劑具有更高的活性,但易流失、難回收的缺點限制了其在實際應用中的推廣,催化劑的應用形式仍是未來的研究重點。
2.3 提高H2O2產量
O2通過2e-ORR生成過氧化氫的過程(式(1),E0=0.70 V vs.RHE)是保證電芬頓技術高效降解有機物的關鍵因素之一,但O2還能通過四電子氧還原反應(4e-ORR)生成H2O(式(6),E0=1.23 V vs.RHE),嚴重影響了H2O2的產量。施加低的應用電位有助于提高H2O2的產量,但伴隨著析氫反應的出現(式(7),E0=0 V vs.RHE),電流效率進一步下降。為了提高H2O2的選擇性生成,往往需要添加助催化劑引導反應向2e-ORR方向進行。

(6)
(7)
碳材料具有較大的比表面積、良好的導電性、容易修飾等優點而被廣泛用作陰極,但是其H2O2選擇性較差。碳材料對H2O2合成的電催化活性和選擇性可以通過改變電子結構來調整[39]。Wang等[40]在碳質載體上原位合成Pd催化劑,H2O2選擇性可達95%左右。除了金屬負載修飾外,非金屬雜原子(O、N、F等)修飾也是常見的方法[41-46]。氧分子的吸附模式對還原途徑影響較大,其中末端吸附即泡林模式更容易通過2e-還原途徑生成H2O2[47-48],羰基(C=O)或羧基(O=C—OH)中正電碳原子可以優先以末端吸附模式吸附氧分子,如圖3所示。在碳中摻雜電負性較高的氮原子,可以通過破壞π共軛體系的完整性和誘導電荷重分布來激活碳π電子,從而改變碳材料的吸附性能,有利于H2O2的生成[45]。Su等[49]證明了引入石墨N可有效促進H2O2的生成。電負性最高的氟在碳納米材料中摻雜后,可以誘導相鄰的碳極化形成活性位點,增強O2與碳的相互作用[50]。Zhao等[51]通過控制F的含量和種類調節對氧還原反應選擇性,與吸附在石墨碳上相比,中間體OOH*吸附在CF2上C的吸附能降低,有利于H2O2的生成。幾種典型的碳基陰極材料及其氧還原活性位點如表4所示。
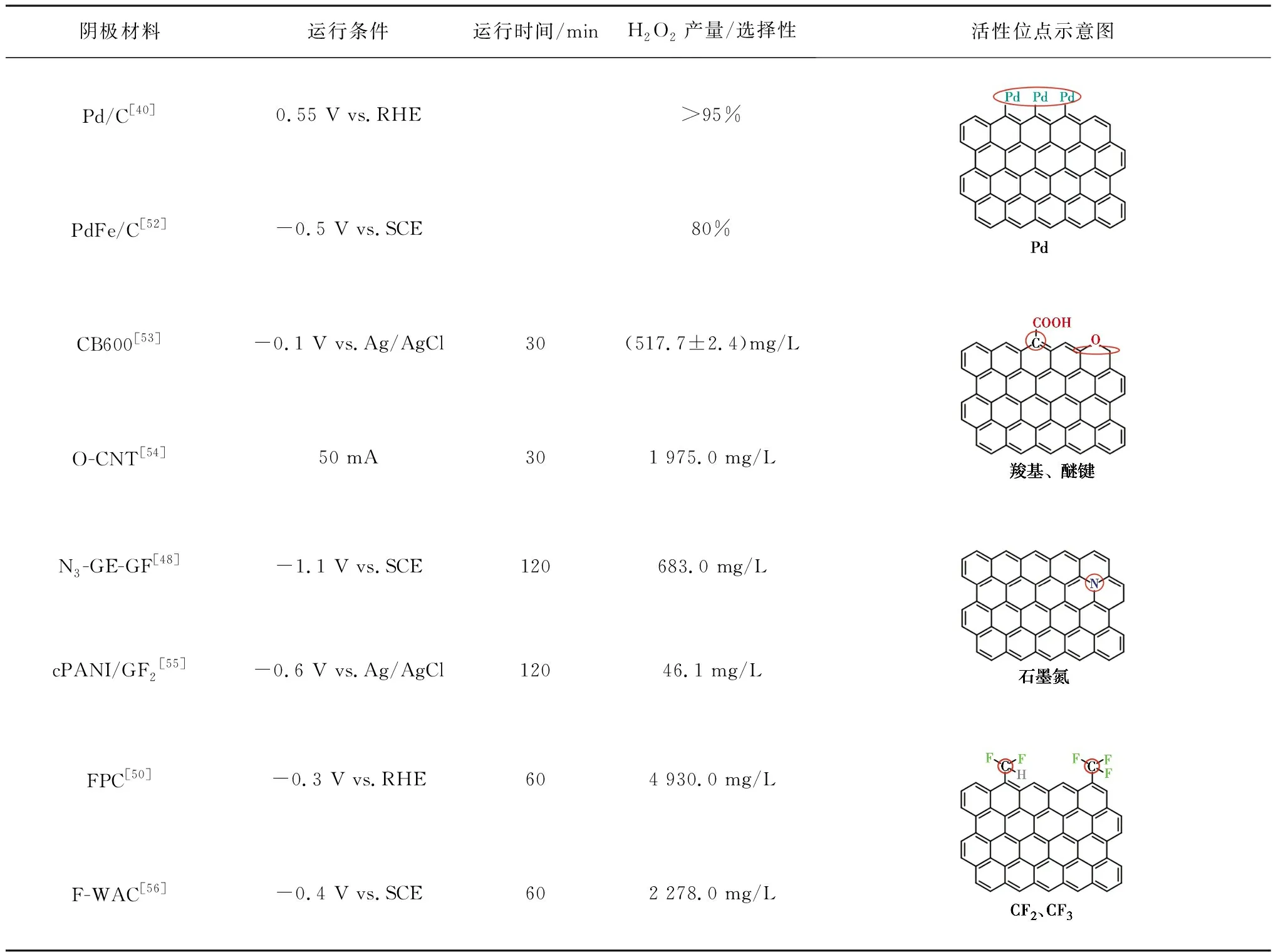
表4 典型碳基陰極材料Table 4 Typical carbon-based cathode material
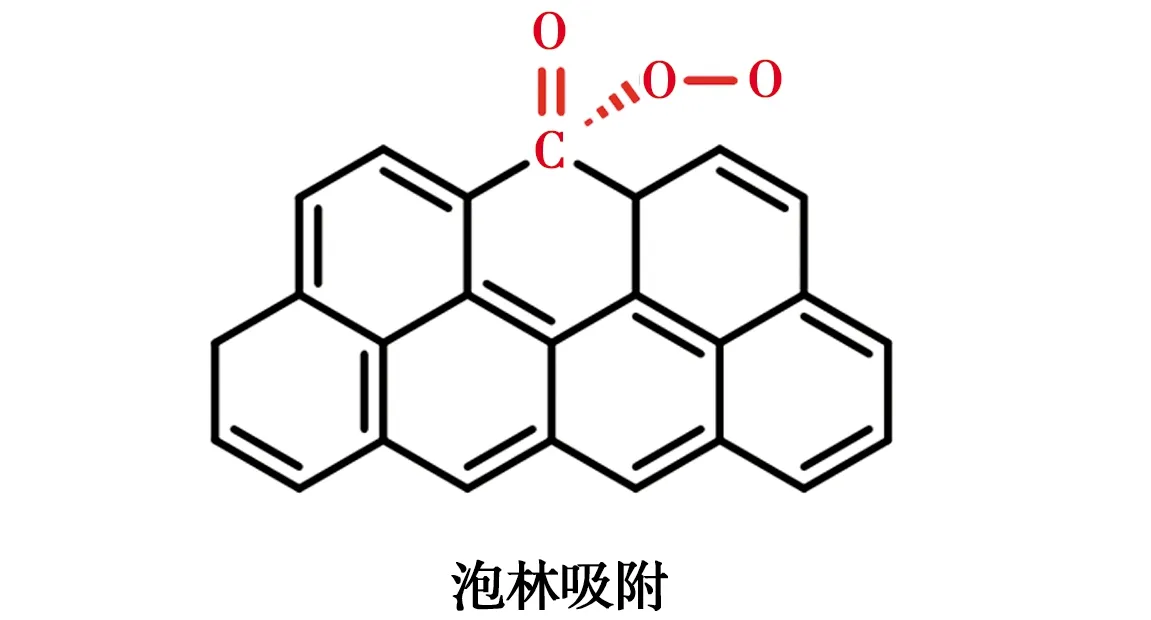
圖3 雙電子還原氧的泡林吸附模型Fig.3 Pauline model of oxygen adsorption toward double
各種改性碳材料被應用于電芬頓體系中。Shen等[52]將鈀鐵合金嵌碳氣凝膠(PdFe /CA)作為陰極降解3-氯酚(3-CP),與鐵碳氣凝膠(Fe/CA)陰極對比發現,Pd的摻雜能夠增強2e-ORR選擇性,PdFe/CA通過(2+1)e-還原途徑先將O2還原為H2O2,然后進一步形成HO·。PdFe /CA陰極電芬頓體系能在不添加外源性H2O2的情況下完全礦化50 mg/L的3-CP。非金屬雜原子摻雜可以避免金屬離子二次泄露問題,Zhang等[53]通過簡單的煅燒法在炭黑表面引入含氧官能團,在-0.1 V vs.Ag/AgCl 的電位下,CB600 FAC(600 ℃煅燒得到的材料)30 min產生(517.7±2.4)mg/L的H2O2,約是未經煅燒處理CB FAC(65.3±5.6)mg/L的8倍,用于電芬頓降解50 mg/L的RhB,2 min內去除率高達91.1%。Cao等[43]在不同溫度下高溫碳化NH2-MIL-88B(Fe)制備了嵌入氧化鐵顆粒的含氮分層多孔碳 (FeOX/NHPCt,t為碳化溫度),在-0.3~-0.8 V vs.SCE電位下,FeOX/NHPC750的H2O2選擇性達到95%~98%,電子轉移數為2.04~2.08,是近似的2e-ORR過程。Lu等[57]以MIL-100 (Fe)∶PANI質量比為2∶1制備了Fe2O3/N-C催化劑,在溶液中檢測出55 mg/L的H2O2,2 h內能完全去除10 mg/L的雙酚A。MOF衍生物保留的孔結構提供了更多的活性位點以及增強傳質過程,有利于高效去除污染物。
由于摻雜O、N、F陰極材料的H2O2選擇性與其形態、含量密切相關,因此,如何有效調控摻雜元素的形態以及比例是未來的研究重點。與此同時,目前已有的調控手段較為復雜,成本大幅增加,開發簡單的合成方法有助于提高實際應用價值。另外,強氧化性環境對催化劑穩定性的影響規律以及對催化位點的失活機制分析仍有待加強。
3 結論與展望
電芬頓技術是一項具有廣闊應用前景的廢水處理技術,無需投加化學試劑,對污染物去除效率高,通過添加助催化劑能改善鐵離子穩定性、加速Fe3+/Fe2+循環以及提高H2O2產量,從而顯著增強難降解有機污染物的去除性能。然而,運行成本高、催化劑穩定性差以及合成工藝復雜、納米催化劑易流失、難以回收等問題仍將限制其大規模應用推廣。助催化劑強化電芬頓技術的改良與發展,仍需側重以下幾個方面:
1)結合計算、表征等手段深入剖析助催化劑強化電芬頓的作用機理以及催化劑的失活機制,進一步解決因催化位點破壞而導致的催化活性降低和二次污染問題。
2)開發簡單、低成本的助催化劑制備方法,并著重研發多功能型助催化劑,兼具兩電子氧還原催化位點和芬頓活性位點,從而簡化電芬頓工藝流程。
3)探究高效穩定的催化劑應用形式與高效穩定型反應器,減少在運行過程中催化劑的損失以及提高重用性,進而降低工程投資和運行成本。
4)聯合電芬頓技術以及其他技術,充分發揮聯合技術氧化還原的性能優勢,實現復合污染物的協同去除。
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
