高效液相色譜法同時測定黃芩莖、葉、花中野黃芩苷含量
2021-09-12 05:23:32王云龍劉春生陳立柱
中國藥業(yè) 2021年17期
王云龍,劉春生,陳立柱,房 岐
(1.黑龍江省雞西市食品藥品檢驗檢測中心,黑龍江 雞西158100;2.北京中醫(yī)藥大學(xué)中藥學(xué)院,北京100102)
黃芩為大宗藥材,源于唇形科植物黃芩Scutellaria baicalensisGeorgi的干燥根,用于治療各種熱證和濕證[1],采收時僅保留地下主根作為藥用部位,地上部分莖、葉、花往往當(dāng)作廢棄物處理,造成大量資源浪費。野黃芩苷是黃芩地上部分的主要成分[2-3],具有抗腫瘤[4]、神經(jīng)保護(hù)[5]、抗炎[6-7]等藥理作用,目前尚無同時測定黃芩莖、葉、花中野黃芩苷含量的研究。本研究中建立了同時測定黃芩莖、葉、花中野黃芩苷含量的高效液相色譜法,為其質(zhì)量標(biāo)準(zhǔn)的制訂和進(jìn)一步開發(fā)利用提供參考。現(xiàn)報道如下。
1 儀器與試藥
1.1 儀器
LC2010 A型高效液相色譜儀(日本島津公司);XPE205型電子天平(德國Mettler Toledo公司,精度為0.01 mg);XM-300UVF型智能靜音超聲波清洗機(小美超聲儀器有限公司,功率為300 W,頻率為40 kHz)。
1.2 試藥
野黃芩苷對照品(上海源葉生物科技有限公司,批號為Y24F11Y16967,純度為98.0%);甲醇(色譜純,德國Merck公司);黃芩莖、葉、花3批樣品均采自黑龍江省雞西市黃芩栽培基地,采集時根據(jù)種植面積平均分為3個區(qū)域,每個區(qū)域隨機采取2株,共6株。樣品采集后,每株分別摘取葉和花,除去雜質(zhì),干燥,得干燥葉和花;莖切段,干燥,得干燥莖。所得樣品經(jīng)雞西市食品藥品檢驗檢測中心楊立志主任藥師鑒定為黃芩的干燥莖、葉和花。樣品來源及相關(guān)信息見表1。

表1 樣品來源及相關(guān)信息Tab.1 Sources and related information of the samples
2 方法與結(jié)果
2.1 色譜條件
色譜柱:Agilent Zorbax SB-C18柱(250 mm×4.6 mm,5 μm);流動相:甲醇(A)-0.4 %磷酸水溶液(B),梯度洗脫(0~15 min時35%A,15~25 min時35%A~80%A,25~32 min時80%A~35%A,32~35 min時35%A);流速:1.0 mL/min;檢測波長:335 nm;柱溫:25℃;進(jìn)樣量:10 μL。
2.2 溶液制備
取野黃芩苷對照品24.88 mg,精密稱定,置100 mL容量瓶中,加70%乙醇溶解并定容,即得質(zhì)量濃度為243.82 μg/mL的對照品貯備液,精密吸取4 mL,置10 mL容量瓶中,用70%乙醇定容,混勻,即得質(zhì)量濃度為97.53 μg/mL的對照品溶液。分別取3號樣品的莖、葉、花,研成細(xì)粉,過4號篩,取莖粉末0.50 g、葉粉末0.13 g、花粉末0.25 g,精密稱定,置50 mL容量瓶中,用70%乙醇定容,密塞,靜置30 min,超聲處理(功率為300 W,頻率為40 kHz)30 min,放冷,用0.45 μm微孔濾膜過濾,即得供試品溶液。取缺黃芩莖、葉、花的陰性樣品,按供試品溶液制備方法制備陰性對照藥材溶液。
2.3 方法學(xué)考察
專屬性試驗:取2.2項下對照品溶液、供試品溶液、陰性對照藥材溶液各適量,按2.1項下色譜條進(jìn)樣測定。結(jié)果供試品溶液色譜圖中,在與對照品溶液色譜相同保留時間處有相應(yīng)色譜峰出現(xiàn),陰性對照無干擾。色譜圖見圖1。
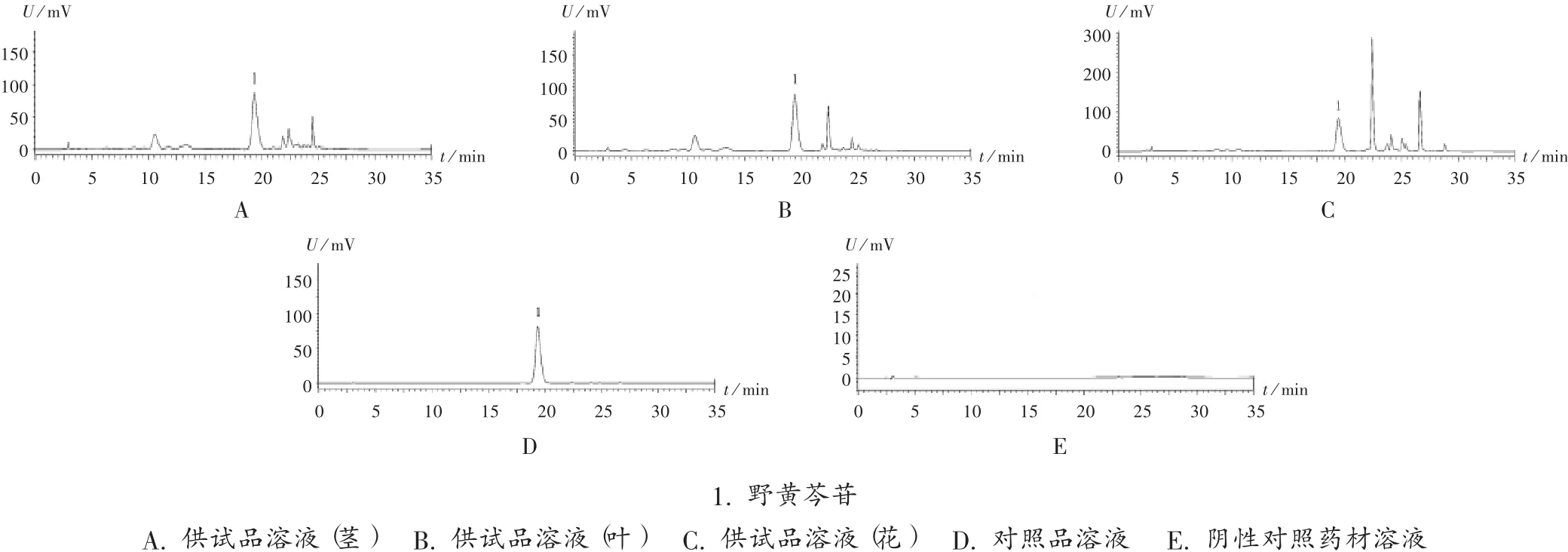
圖1 高效液相色譜圖1.scutellarinA.Test solution(stem)B.Test solution(leaf)C.Test solution(flower)D.Reference solution E.Negative reference medicinal material solutionFig.1 HPLC chromatograms
線性關(guān)系考察:精密吸取2.2項下對照品貯備液1,2,4,6,8,10 mL,分別置10 mL容量瓶中,用70%乙醇定容,搖勻,得系列質(zhì)量濃度的混合對照品溶液,按2.1項下色譜條件進(jìn)樣測定,以峰面積(Y)為縱坐標(biāo)、對照品溶液質(zhì)量濃度(X,μg/mL)為橫坐標(biāo)進(jìn)行線性回歸,得回歸方程Y=25 881X+93 582(r=0.999 9,n=6)。結(jié)果表明,野黃芩苷質(zhì)量濃度在24.38~243.80 μg/mL范圍內(nèi)與峰面積線性關(guān)系良好。
精密度試驗:取黃芩葉(3號樣品),依法制備供試品溶液,按2.1項下色譜條件連續(xù)進(jìn)樣測定6次。結(jié)果野黃芩苷峰面積的RSD為0.31%(n=6),表明儀器精密度良好。
重復(fù)性試驗:取黃芩葉(3號樣品)0.13 g,精密稱定,共6份,依法制備供試品溶液,按2.1項下色譜條件進(jìn)樣測定。結(jié)果野黃芩苷含量的RSD為1.53%(n=6),表明方法重復(fù)性良好。
穩(wěn)定性試驗:取黃芩葉(3號樣品),依法制備供試品溶液,分別于0,2,4,8,16,24,48 h時按2.1項下色譜條件進(jìn)樣測定。結(jié)果野黃芩苷峰面積的RSD為0.92%(n=7),表明供試品溶液在48 h內(nèi)穩(wěn)定。
加樣回收試驗:取已知含量的黃芩葉(3號樣品)粉末65 mg,精密稱定,共6份,分別置50 mL容量瓶中,精密加入2.2項下對照品貯備液10 mL,用70%乙醇定容,依法制備供試品溶液,按2.1項下色譜條件進(jìn)樣測定,并計算加樣回收率。結(jié)果見表2。
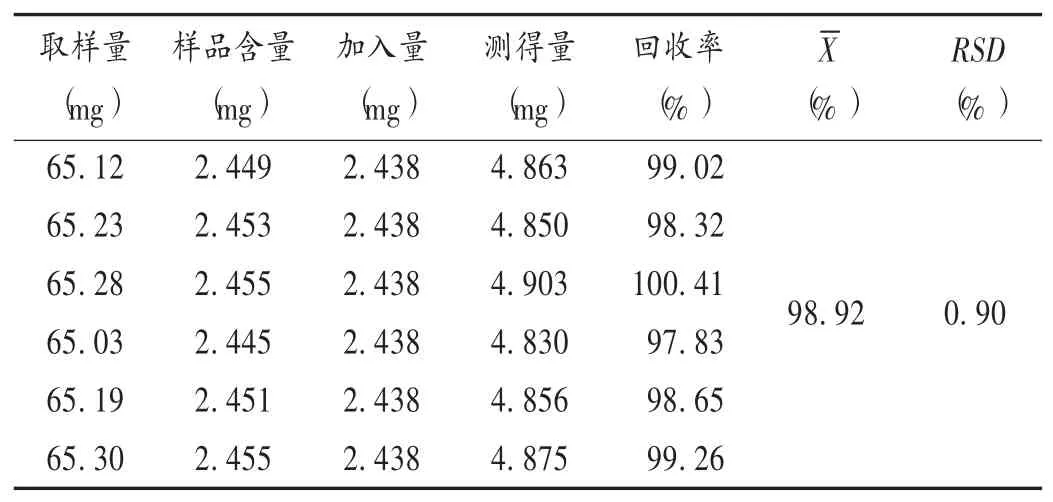
表2 黃芩葉中野黃芩苷加樣回收試驗結(jié)果(n=6)Tab.2 Results of the recovery test of scutellarin in the leaves of Scutellaria baicalensis(n=6)
2.4 樣品含量測定
分別取3批黃芩莖、葉、花,各3份,依法制備供試品溶液,按2.1項下色譜條件進(jìn)樣測定,采用外標(biāo)法計算野黃芩苷的含量,結(jié)果見表3。可見,黃芩莖、葉、花中野黃芩苷含量由高至低依次為葉、花、莖,葉中野黃芩苷的含量為莖或花的2~3倍。3批樣品中野黃芩苷含量差異不大,表明生長年限對黃芩莖、葉、花中野黃芩苷含量的影響不顯著。

表3黃芩莖、葉、花中野黃芩苷含量測定結(jié)果(%,n=3)Tab.3 Content determination of scutellarin in the stems,leaves and flowers of Scutellaria baicalensis(%,n=3)
3 討論
在前期試驗中發(fā)現(xiàn),采用高效液相色譜法測定時,黃芩莖、葉、花中野黃芩苷色譜峰的信號較強,故選擇野黃芩苷為檢測指標(biāo)。考察了不同提取方法(70%乙醇超聲提取、70%乙醇回流提取、甲醇超聲提取、甲醇回流提取)對含量測定結(jié)果的影響,結(jié)果以70%乙醇為提取溶劑時,先靜置30 min,再超聲處理30 min,黃芩莖、葉、花中野黃芩苷的含量最高,考慮超聲提取法簡便,故選擇70%乙醇超聲提取。通過掃描紫外光譜發(fā)現(xiàn),在335 nm波長下有最大吸收,故選擇335 nm為檢測波長。
前期試驗中發(fā)現(xiàn),不同樣品黃芩葉含量差異較大,考慮其化學(xué)成分可能受生長時間和不同生長部位等因素影響,故僅選擇黃芩葉樣品進(jìn)行方法學(xué)考察。黃酮類化合物多顯弱酸性,易解離,影響分離效果,采用離子抑制色譜法加入一定量磷酸,通過梯度洗脫,各色譜峰分離較好。
測定樣品含量時發(fā)現(xiàn),葉中野黃芩苷的含量為莖或花的2~3倍,提示今后制訂黃芩莖、葉、花的質(zhì)量標(biāo)準(zhǔn)時,應(yīng)充分考慮莖、葉、花的質(zhì)量比例。生長年限對黃芩莖、葉、花野黃芩苷含量的影響不顯著,其原因可能與每年秋季對莖、葉、花割秧或焚燒處理使次年發(fā)新枝葉有關(guān)。
綜上所述,本方法操作簡便,專屬性強,準(zhǔn)確度高,重復(fù)性好,可用于黃芩莖、葉、花中野黃芩苷的含量測定,可為今后黃芩地上部分質(zhì)量標(biāo)準(zhǔn)的制訂和進(jìn)一步開發(fā)利用提供依據(jù)。
