小兒解表口服液HPLC特征圖譜建立及4種成分測定
2021-09-24 11:10:22解盈盈翟士旭林永強
中成藥 2021年9期
汪 冰, 解盈盈, 薛 菲, 翟士旭, 林永強*
(1.山東省食品藥品檢驗研究院,山東省中藥標準創新與質量評價工程實驗室,山東 濟南 250101;2.威海人生藥業集團股份有限公司,山東 濟南 250022)
小兒解表口服液由金銀花、葛根、牛蒡子、黃芩、連翹、蒲公英、紫蘇葉等藥材經提取而成,具有宣肺解表,清熱解毒之功效,臨床主要用于治療小兒外感風熱引起的惡寒發熱、頭痛咳嗽、鼻塞流涕、咽喉痛癢[1-4]。該制劑現行質量控制標準主要是連翹苷、黃芩苷、葛根素的TLC定性鑒別和金銀花中綠原酸定量測定[5],雖然在一定程度上保障了產品質量,但無法全面反映其質量狀況。因此,本實驗建立小兒解表口服液HPLC特征圖譜,并對隱綠原酸、葛根素、黃芩苷、牛蒡苷含量進行測定,以期從整體上評價該制劑質量[6-15]。
1 材料
Waters 2695高效液相色譜儀(美國Waters公司,配置二極管陣列檢測器、Empower 2色譜工作站);CP225D電子分析天平(德國賽多利斯公司);Milli-Q 超純水系統(美國默克密理博公司);KQ-500DB型數控超聲波清洗器(功率500 W,昆山市超聲儀器有限公司)。小兒解表口服液13批,購自威海人生藥業集團股份有限公司,批號分別為20161201(S1)、20161202(S2)、20161203(S3)、0190101(S4)、0190102(S5)、0190103(S6)、0190104(S7)、0190201(S8)、0190202(S9)、0190203(S10)、0190301(S11)、0190302(S12)、0190303(S13)。
綠原酸(批號110753-201314,純度96.6%)、黃芩苷(批號110715-201117,純度91.7%)、葛根素(批號110752-201313,純度95.5%)、連翹苷(批號110821-201615,純度94.9%)、牛蒡苷(批號110819-200606)、4,5-二-O-咖啡酰奎寧酸(批號111894-201102)、咖啡酸(批號110885-200102)對照品均購自中國食品藥品檢定研究院;隱綠原酸對照品(批號MUST-19032403,純度99.07%)購自成都普思生物科技有限公司。甲醇、乙腈為色譜純;其他試劑均為分析純;水為Millipore純化水。
2 方法與結果
2.1 色譜條件 Agilent Extend-C18色譜柱(250 mm×4.6 mm,5 μm);流動相乙腈(A)-0.4%磷酸(B),梯度洗脫(0~10 min,10%A;10~35 min,10%~25%A;35~40 min,25%~35%A;40~43 min,35%A);體積流量1.0 mL/min;柱溫35 ℃;檢測波長230 nm;進樣量10 μL。
2.2 溶液制備
2.2.1 對照品溶液 精密稱取各對照品適量,50%甲醇制成每1 mL分別含綠原酸45.24 μg、隱綠原酸33.00 μg、咖啡酸5.04 μg、葛根素20.38 μg、4,5-二-O-咖啡酰奎寧酸41.56 μg、黃芩苷97.03 μg、連翹苷14.80 μg、牛蒡苷69.63 μg的溶液,即得。
2.2.2 對照藥材溶液 取蒲公英、金銀花、紫蘇葉、葛根、黃芩、連翹、牛蒡子對照藥材各0.1 g,25 mL 50%甲醇超聲處理30 min,濾過,取續濾液,即得。
2.2.3 供試品溶液 精密量取口服液2 mL,置于50 mL量瓶中,50%甲醇稀釋至刻度,搖勻,濾過,取續濾液,即得。
2.2.4 陰性樣品溶液 按制劑處方和工藝,分別制備缺蒲公英、缺金銀花、缺葛根、缺黃芩、缺連翹、缺牛蒡子、缺紫蘇葉陰性樣品,按“2.2.3”項下方法制備,即得。
2.3 特征圖譜方法學考察
2.3.1 精密度試驗 取樣品(S1),按“2.2.3”項下方法制備供試品溶液,在“2.1”項色譜條件下進樣測定6次,所得色譜圖中1~8號峰以綠原酸為參照,9~13號峰以黃芩苷為參照,測得13個共有峰相對保留時間、相對峰面積RSD分別小于0.5%、1.0%,表明儀器精密度良好。
2.3.2 重復性試驗 取樣品(S1),按“2.2.3”項下方法平行制備6份供試品溶液,在“2.1”項色譜條件下進樣測定,所得色譜圖中1~8號峰以綠原酸為參照,9~13號峰以黃芩苷為參照,測得13個共有峰相對保留時間、相對峰面積RSD均分別小于0.5%、1.5%,表明該方法重復性良好。
2.3.3 穩定性試驗 取樣品(S1),按“2.2.3”項下方法制備供試品溶液,于0、5、10、15、20、25 h在“2.1”項色譜條件下進樣測定,所得色譜圖中1~8號峰以綠原酸為參照,9~13號峰以黃芩苷為參照,測得13個共有峰相對保留時間、相對峰面積RSD均分別小于0.5%、1.0%,表明溶液在25 h內穩定性良好。
2.4 圖譜生成 按“2.2.3”項下方法制備13批樣品的供試品溶液,在“2.1”項色譜條件下進樣測定,采用“中藥色譜指紋圖譜相似度評價系統”(2012年版)對其色譜圖進行分析(圖1),選擇重復性、分離度較好,可歸屬到處方藥味的13個色譜峰作為共有峰。由于共有峰在圖譜中主要分布于前后部分,較為分散,故以保留時間適中、峰面積較大、分離度較好的2號峰(綠原酸)、10號峰(黃芩苷)為參照,保留時間在25 min前的1~8號峰選擇前者,25 min后的9~13號峰選擇后者,計算共有峰的相對保留時間和相對峰面積,結果見表1~2。
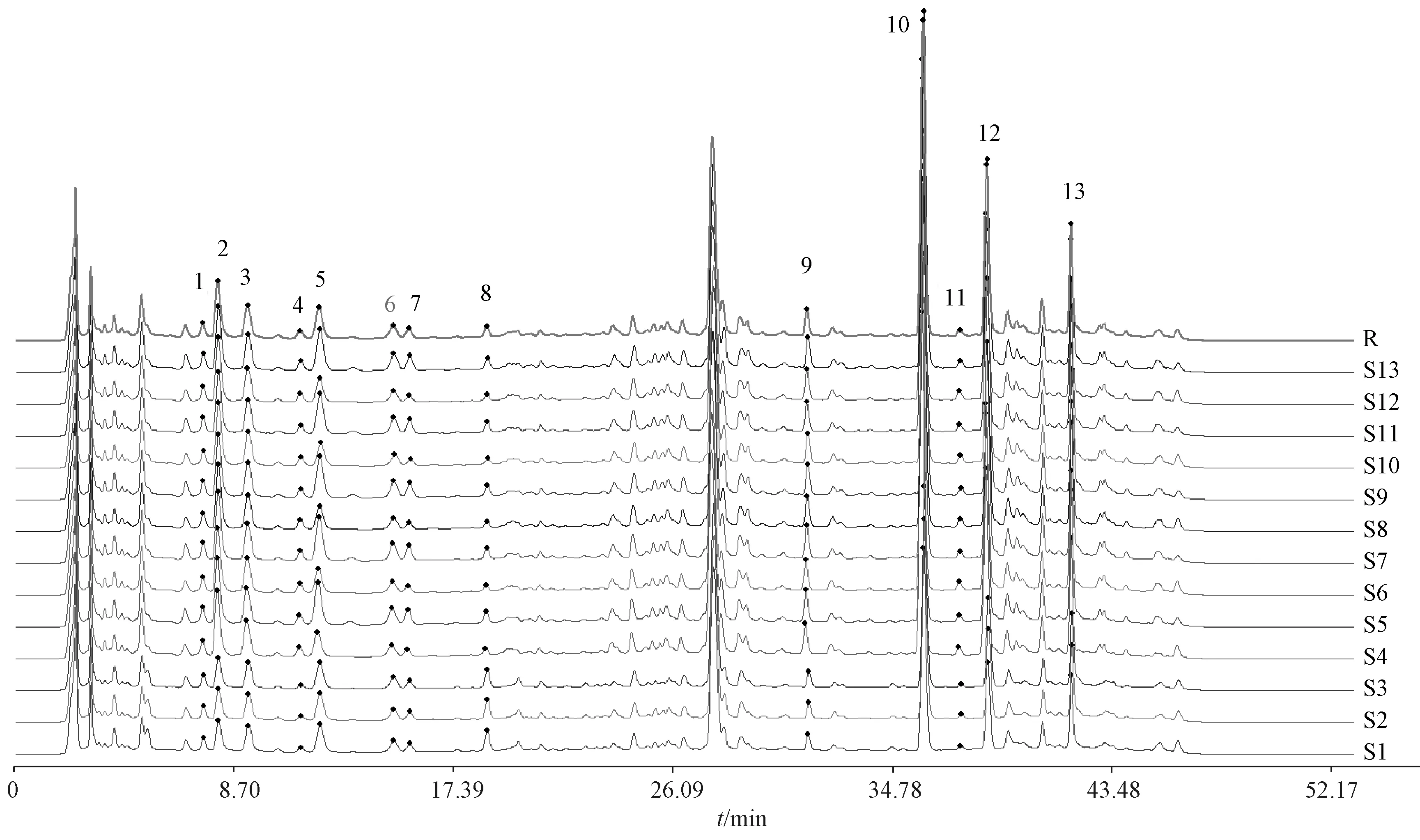
圖1 13批樣品HPLC特征圖譜Fig.1 Characteristic HPLC chromatograms of thirteen batches of samples
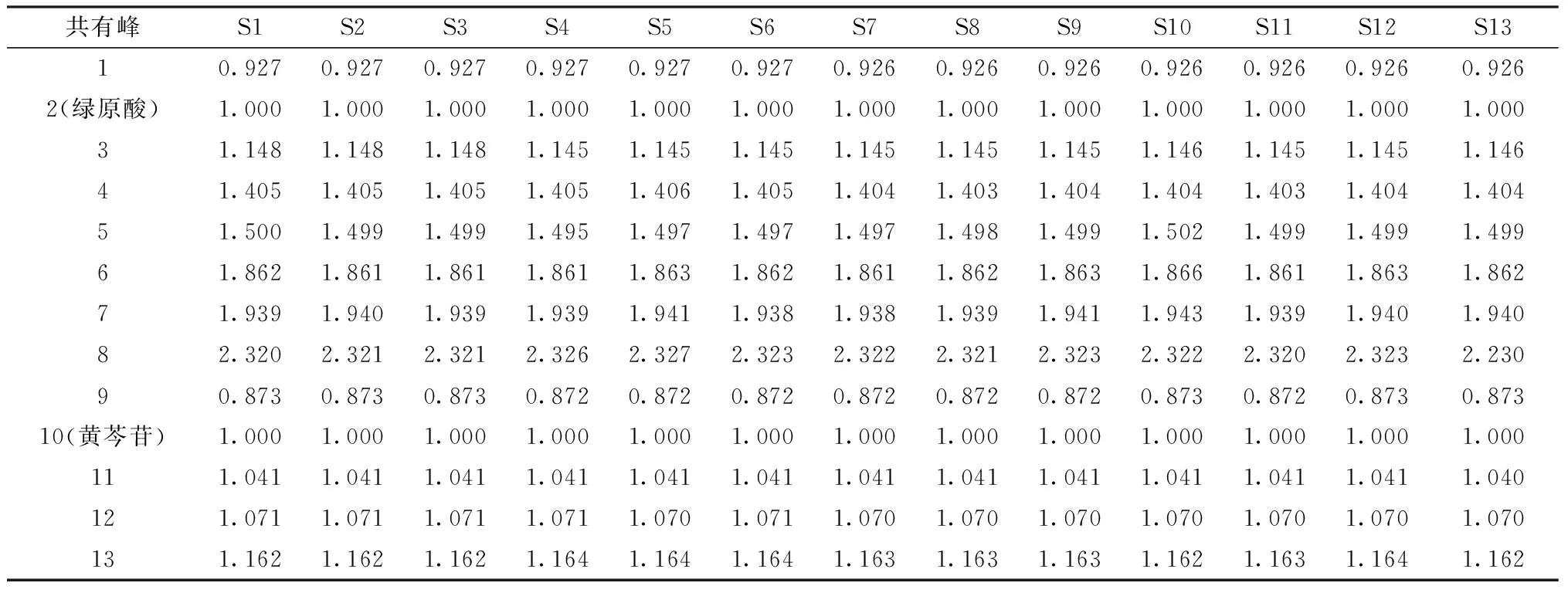
表1 共有峰相對保留時間

表2 共有峰相對峰面積
2.5 共有峰相對保留時間規定值確定 以綠原酸為參照峰1,計算前8個共有峰的相對保留時間;以黃芩苷為參照峰2,計算后5個共有峰的相對保留時間。以13批樣品HPLC特征圖譜中13個共有峰的相對保留時間平均值為規定值,分別為0.93(峰1)、1.00(峰2)、1.15(峰3)、1.40(峰4)、1.50(峰5)、1.86(峰6)、1.94(峰7)、2.32(峰8)、0.87(峰9)、1.00(峰10)、1.04(峰11)、1.07(峰12)、1.16(峰13),并且偏差均應在±5%以內。由表2可知,8個共有峰相對峰面積RSD較大(>10%),故只規定共有峰相對保留時間,而不規定其相對峰面積。
2.6 共有峰指認及歸屬 通過與對照藥材溶液、陰性樣品溶液色譜峰進行比較,可判定1號色譜峰來自蒲公英,2號色譜峰為金銀花、蒲公英、牛蒡子的共有峰,3號色譜峰來自金銀花,4號色譜峰為蒲公英、紫蘇葉的共有峰,5~8號色譜峰來自葛根,9號色譜峰為金銀花、蒲公英的共有峰,10、13號色譜峰來自黃芩,11號色譜峰來自連翹,12號色譜峰來自牛蒡子。再進行對照品定位及紫外光譜比較,確定峰2為綠原酸,峰3為隱綠原酸,峰4為咖啡酸,峰5為葛根素,峰9為4,5-二-O-咖啡酰奎寧酸,峰10為黃芩苷,峰11為連翹苷,峰12為牛蒡苷。色譜圖見圖2。
2.7 計量學分析
2.7.1 相似度計算 將13批樣品數據導入中藥色譜指紋圖譜相似度評價系統,以對照特征圖譜為參照,進行共有峰匹配后計算相似度,結果見表3,可知均高于0.98,表明各批樣品之間差異較小。
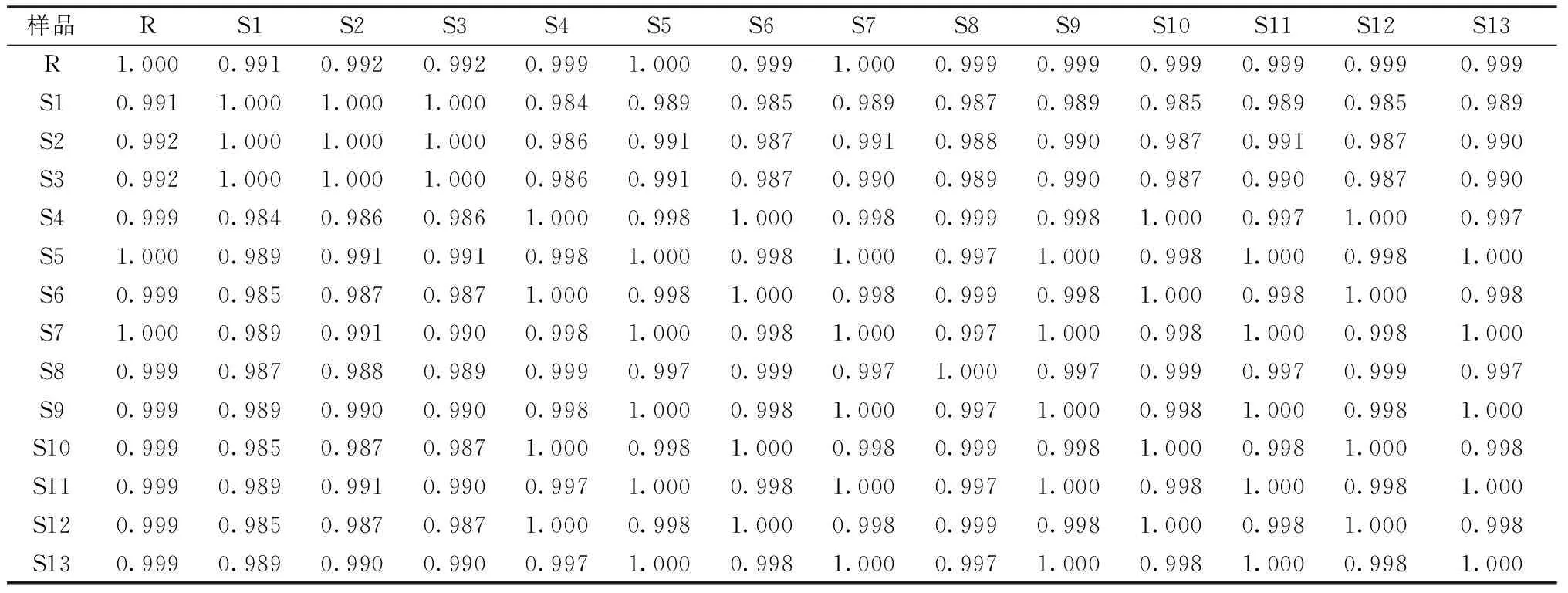
表3 13批樣品相似度

2.綠原酸 3.隱綠原酸 4.咖啡酸 5.葛根素 9.4,5-O-二咖啡酰奎寧酸 10.黃芩苷 11.連翹苷 12.牛蒡苷2.chlorogenic acid 3.cryptochlorogenic acid 4.caffeic acid 5.puerarin 9.isochlorogenic acid C 10.baicalin 11.phillyrin 12. arctiin圖2 各成分HPLC色譜圖Fig.2 HPLC chromatograms of various constituens
2.7.2 聚類分析 通過SPSS19.0軟件,以13個共有峰峰面積為變量,采用組間聯接系統聚類法中的歐氏距離對13批樣品進行聚類分析,結果見圖3。由此可知,各批樣品大致分為2類,S1~S3聚為一類,生產日期為2016年;S4~S13聚為另一類,生產日期為2019年。
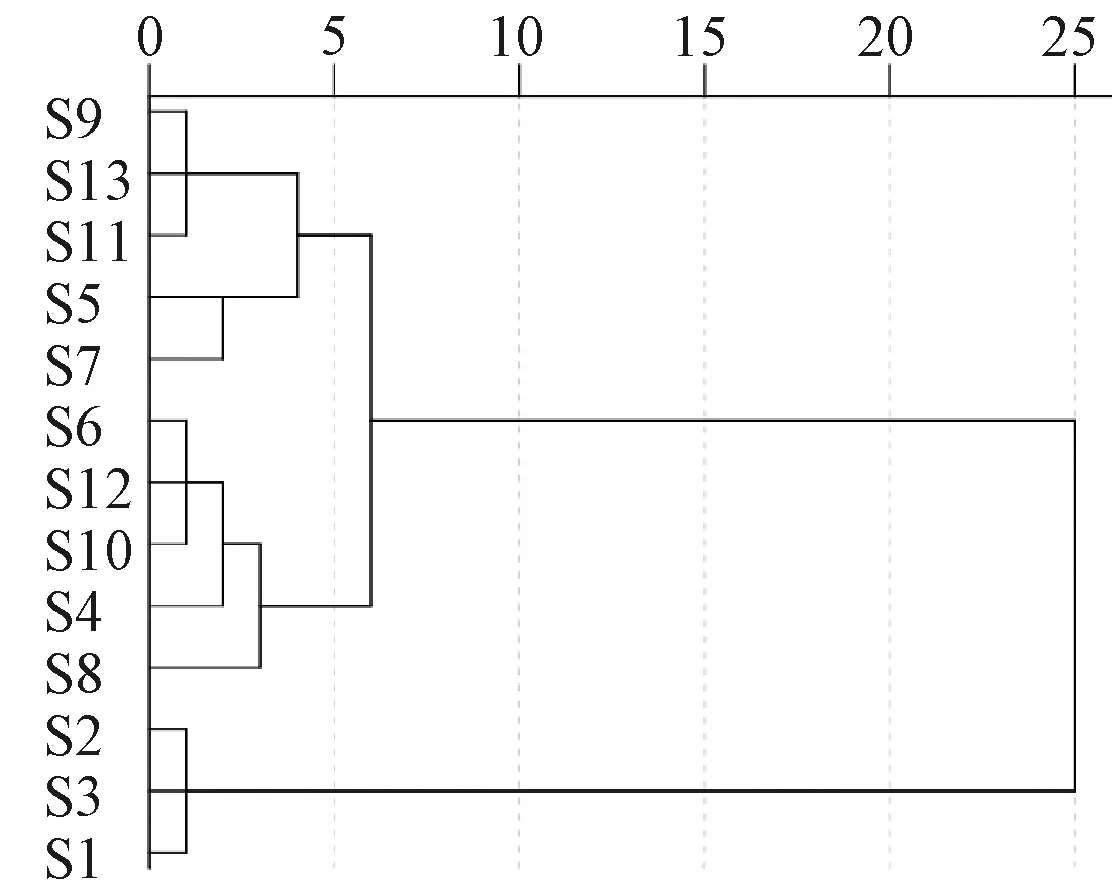
圖3 13批樣品聚類分析圖Fig.3 Cluster analysis plot for thirteen batches of samples
2.8 各成分含量測定
2.8.1 溶液制備
2.8.1.1 對照品溶液 精密稱取各對照品,50%甲醇制成每1 mL分別含牛蒡苷0.208 89 mg、黃芩苷0.291 09 mg、葛根素0.030 57 mg、隱綠原酸0.099 0 mg的溶液,即得。
2.8.1.2 供試品溶液 精密量取口服液2 mL,置于50 mL量瓶中,50%甲醇稀釋至刻度,搖勻,濾過,取續濾液,即得。
2.8.2 方法學考察
2.8.2.1 線性關系考察 取“2.8.1.1”項下對照品溶液,50%甲醇稀釋(牛蒡苷0.013 93、0.034 82、0.069 63、0.104 45、0.139 26、0.174 08、0.208 89 mg/mL,黃芩苷0.019 41、0.048 52、0.097 03、0.145 55、0.194 06、0.242 58、0.291 09 mg/mL,葛根素0.002 04、0.005 10、0.010 19、0.015 29、0.020 38、0.025 48、0.030 57 mg/mL,隱綠原酸0.006 6、0.016 5、0.033 0、0.049 5、0.066 0、0.082 5、0.099 0 mg/mL),在“2.1”項色譜條件下進樣測定。以對照品質量濃度為橫坐標(X),峰面積為縱坐標(Y)進行回歸,得方程分別為牛蒡苷Y=1.768 0×107X-5.531 9×104(r=0.999 9)、黃芩苷Y=1.731 1×107X-4.899 7×104(r=1.000 0)、葛根素Y=3.038 2×107X-1.910 9×104(r=1.000 0)、隱綠原酸Y=1.399 1×107X-2.297 0×104(r=0.999 9),分別在0.013 93~0.208 89、0.019 41~0.291 09、0.002 04~0.030 57、0.006 6~0.099 0 mg/mL范圍內線性關系良好。
2.8.2.2 精密度試驗 取“2.8.2.1”項下各成分第3個質量濃度的對照品溶液,在“2.1”項色譜條件下進樣測定6次,測得牛蒡苷、黃芩苷、葛根素、隱綠原酸峰面積RSD分別為1.16%、0.92%、0.89%、1.03%,表明儀器精密度良好。
2.8.2.3 重復性試驗 取樣品(S1),按“2.8.1.2”項下方法平行制備6份供試品溶液,在“2.1”項色譜條件下進樣測定,測得牛蒡苷、黃芩苷、葛根素、隱綠原酸含量RSD分別為1.10%、0.37%、0.23%、0.66%,表明該方法重復性良好。
2.8.2.4 穩定性試驗 取供試品溶液(S1),于0、5、10、15、20、25 h在“2.1”項色譜條件下進樣測定,測得牛蒡苷、黃芩苷、葛根素、隱綠原酸峰面積RSD分別為0.86%、0.12%、0.42%、1.12%,表明溶液在25 h內穩定性良好。
2.8.2.5 加樣回收率試驗 取各成分含量已知的樣品6份(S1),按“2.8.1.2”項下方法平行制備6份供試品溶液,每份精密量取1 mL至50 mL量瓶中,精密加入對照品溶液[牛蒡苷(0.208 9 mg/mL)5 mL、黃芩苷(0.242 6 mg/mL)10 mL、葛根素(0.101 9 mg/mL)3 mL、隱綠原酸(0.099 0 mg/mL)5 mL],50%甲醇定容至刻度,搖勻,濾過,取續濾液,在“2.1”項色譜條件下進樣測定,計算回收率,結果見表4。

表4 各成分加樣回收率試驗結果(n=6)
2.8.3 樣品含量測定 取13批樣品,按“2.8.1.2”項下方法制備供試品溶液,在“2.1”項色譜條件下各進樣10 μL測定,外標法計算含量,結果見表5。
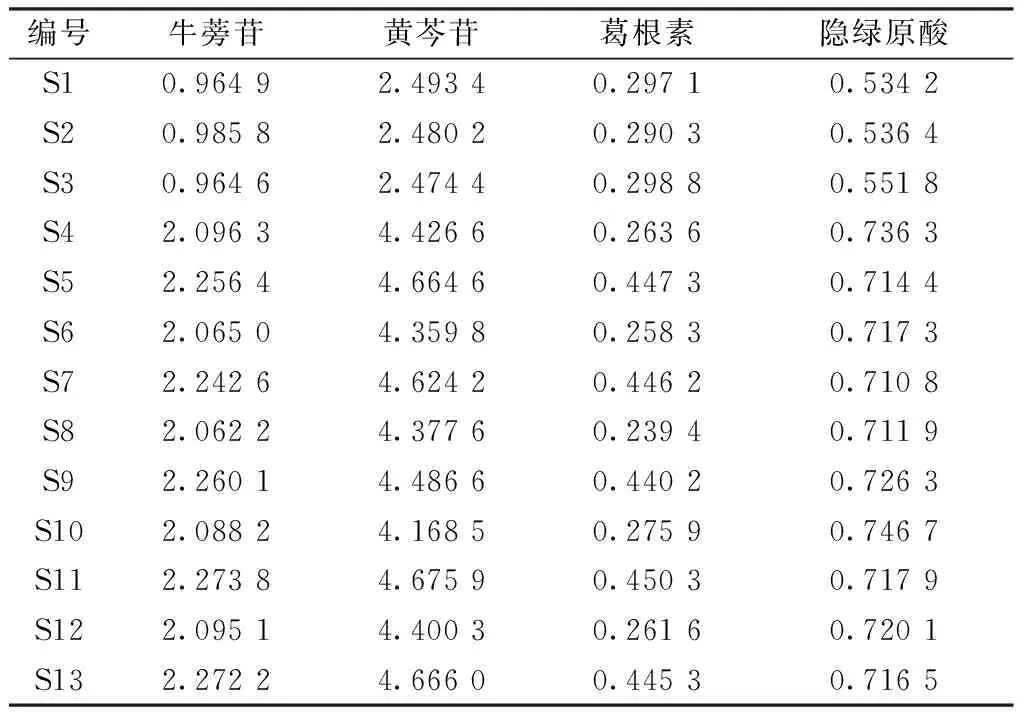
表5 各成分含量測定結果(mg/mL,n=3)
3 討論
3.1 檢測波長選擇 本實驗采用二極管陣列檢測器對8種成分的紫外吸收光譜進行掃描,發現在230 nm下色譜峰數目較多,而且除黃芩苷外均有較好的吸收;該處波長雖為黃芩苷較低吸收,但可減少與其他色譜峰的差異,同時各成分色譜峰峰形、分離度均較好。因此,選擇230 nm作為檢測波長。
3.2 耐用性考察 本實驗考察了4種色譜柱(Agilent Extend-C18,5 μm,250 mm×4.6 mm;依利特Hypersil BDS C18,5 μm,250 mm×4.6 mm;Thermo BDS Hypersil C18,5 μm, 250 mm×4.6 mm;Thermo ODS C18,5 μm,250 mm×4.6 mm),其中Agilent Extend、Thermo BDS Hypersil色譜柱重復性較好。然后,對新購買的、使用不同年限的色譜柱進行,發現其重復性均較好。
3.3 HPLC特征圖譜分析 對13批小兒解表口服液進行聚類分析,發現不同生產年限樣品分別聚為一類,推測可能是由于原料受生長環境、采收時間、加工貯存等因素的影響;另一方面,口服液長時間放置容易出現沉淀,會帶來穩定性方面的問題,隱綠原酸等酚酸類成分易分解轉化[16-17],而黃芩苷等黃酮類成分溶解性較差,也可能影響穩定性。另外,相同生產年限的不同批次樣品雖然個別共有峰略有差異,但其質量基本穩定。
4 結論
本實驗建立了小兒解表口服液HPLC特征圖譜,確定了13個共有峰,通過與對照藥材比對歸屬了方中7個藥味,再與對照品比對指認了其中8種成分,發現13批樣品相似度均大于0.98,表明特征圖譜能較全面反映制劑整體狀況。同時,建立了專屬性較強的牛蒡苷、黃芩苷、葛根素、隱綠原酸含量測定方法,可為小兒解表口服液的質量控制提供參考依據。
