芪參地黃顆粒質(zhì)量標(biāo)準(zhǔn)的建立
2021-10-05 10:41:08王藝璇成光宇張洪銘解生旭鄭明善
吉林中醫(yī)藥 2021年9期
王藝璇,孫 堯,王 健,成光宇,張洪銘,解生旭,鄭明善*
(1.延邊大學(xué)生物教研室,吉林 延吉 136200;2.長春中醫(yī)藥大學(xué)附屬醫(yī)院實驗中心,長春 130117;3.吉林省中醫(yī)藥科學(xué)院植化室,長春 130012)
“芪參地黃顆粒”為擬研制開發(fā)的治療重癥肌無力的中藥復(fù)方新藥,由我院王健教授提出,他本人擔(dān)任吉林省高校“創(chuàng)新團(tuán)隊發(fā)展計劃”團(tuán)隊帶頭人,長白山學(xué)者特聘教授等。王健教授將學(xué)術(shù)與臨床經(jīng)驗相結(jié)合,別具一格的以“稟賦”和“虛損”立論指出痿證的病機關(guān)鍵,提出了病機關(guān)鍵為“脾腎虧虛,腦髓不足”,治療方法為“健脾益氣補髓法”。制劑由黃芪、黨參等十七味中藥材組成。方中黃芪、黨參為君藥,具有益氣壯陽、健脾潤肺的功效,共奏扶正補脾益氣之功;白術(shù)、枸杞子、巴戟天、黃精、山藥助君藥補氣健脾補血生津,生地黃、狗脊、炙甘草祛風(fēng)除濕,以上八者共為臣藥;當(dāng)歸具有養(yǎng)血和營,協(xié)君藥補氣養(yǎng)血,地龍、鉤藤二藥合用增強熄風(fēng)通絡(luò)之功,陳皮調(diào)中、燥濕、痰濕使藥物補而不滯,五者共為佐藥;柴胡、升麻、葛根三者相配伍,協(xié)助君藥以升提下陷之中氣,擔(dān)任佐使。本實驗運用TLC 法對QSDH 中黃芪、黨參、枸杞子、狗脊、當(dāng)歸、陳皮、葛根進(jìn)行鑒別,并用HPLC 法測定黃芪甲苷的含量來控制QSDH 的質(zhì)量,為其質(zhì)量標(biāo)準(zhǔn)的制定奠定基礎(chǔ)。
1 儀器與材料
LC-2010C型高效液相色譜儀(日本島津公司);KQ -300DE型數(shù)控超聲波清洗器(昆山市超聲儀器有限公司);分析天平萬分之一FA2004N 天平 YUEPING 上海越平科學(xué)儀器(蘇州制造有限公司);十萬分之一 METTLER TOLEDO AG245 瑞士;GOODLOOK-100型薄層色譜成像系統(tǒng)(上海科哲生化科技有限公司);Milli -Q(MERC KMILLIPORE);蒸發(fā)光散射檢測器(ELSD,Alltech)。對照品:黃芪甲苷(批號:110781-20166,97.4%)、橙皮苷(批號:110721-201617,96.1%)、葛根素(批號:110752-201816,95.4%)以上對照品均來自于中國食品藥品檢定研究院。芪參地黃顆粒(批號:20191201/20191202/20191203),吉林省中醫(yī)藥科學(xué)院的制劑室提供;HPLC 定量分析中使用的乙腈、水均為色譜級,TLC 定性實驗中使用到的化學(xué)試劑均為分析級。
2 方法與結(jié)果
2.1 薄層鑒別
2.1.1 黃芪鑒別 取QSDH 約3 g,緩緩加入20 mL量的甲醇,加熱并用冷凝管回流1 h,得到的濾液加于100~120 目的中性氧化鋁柱(5 g,內(nèi)徑為10~15 mm)上,用40%甲醇洗脫,用量為100 mL,將洗脫液收集,然后蒸干,用30 mL 的水使蒸干物溶解,拿20 mL 量的正丁醇(水飽和)搖晃2次,歸并正丁醇液再用水洗滌2 次(每次20 mL),留下正丁醇液蒸干,蒸發(fā)后的干物質(zhì)滴0.5 mL 甲醇使其溶解,成為供試品溶液。拿一些量的黃芪甲苷對照品,與甲醇混合制成1 mL/mg 溶液,作對照品溶液。按照薄層色譜法試驗[1],在同一塊高效G 上點2 種溶液每種4 μL,用甲醇-三氯甲烷-水(8:13:2)的下層溶液展開,拿出,揮干后噴1%香草醛硫酸乙醇溶液,105℃下加熱,結(jié)果顯示斑點清晰可見。被測顆粒色譜中,白光下顯示和對照品色譜同位置上一樣的藍(lán)紫色斑點,見圖1。
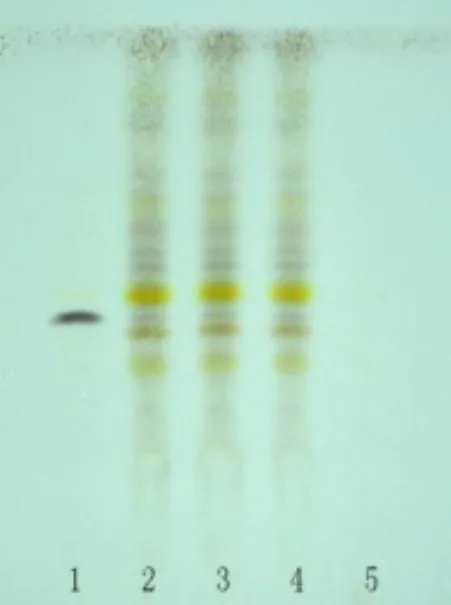
圖1 黃芪 TLC 鑒別
2.1.2 黨參鑒別 取QSDH約1 g,加25 mL量的甲醇,經(jīng)過30 min 的超聲處理,濾過,干燥,殘渣用15 mL量的水溶解,過 D101型大孔吸附樹脂柱(內(nèi)徑為1.5 cm,柱高為10 cm),拿50 mL 量的水洗脫,丟棄水液,再加50 mL50%濃度的乙醇洗脫,混合洗脫液,蒸發(fā)干,滴1 mL 的甲醇溶解,配成供試溶液。另取黨參對照藥材1 g,用上面方法配成對照藥材溶液。按照薄層色譜法試驗[1],在同一塊高效G 上點2 種溶液,每種5 μL,用甲醇-三氯甲烷-水(7:13:2)的下層溶液展開,拿出,放干,噴現(xiàn)配的1:1 的顯色劑2%三氯化鐵溶液-1%鐵氰化鉀溶液。被測顆粒色譜中,白光下顯示和對照品色譜同位置上一樣顏色的斑點,見圖2。

圖2 黨參 TLC 鑒別
2.1.3 枸杞子鑒別 取QSDH 約1 g,加35 mL 量的水,經(jīng)過15 min 加熱煮沸,放冷,濾過,加15 mL 量的乙酸乙酯振搖提取,留下乙酸乙酯液,濃縮成1 mL 作供試品溶液。另拿枸杞子對照藥材1 g,用上面方法配對照藥材標(biāo)準(zhǔn)溶液。按照薄層色譜法試驗[1],在同一塊高效G 上點2 種溶液,每種5 μL,用乙酸乙酯-三氯甲烷-甲酸(3:2:1)展開,拿出,揮干,被測顆粒色譜中,紫外光燈(365 nm)下顯示和對照藥材色譜同位置上一樣顏色的熒光斑點,見圖 3。
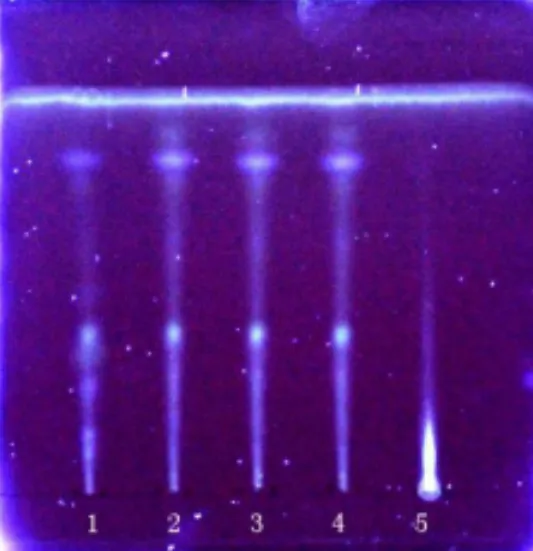
圖3 枸杞子 TLC 鑒別
2.1.4 狗脊鑒別 取QSDH 約2 g,加50 mL 量的甲醇后經(jīng)超聲處理30 min,濾過,蒸干,滴1 mL 甲醇溶成供試品溶液。另取狗脊對照藥材粉末2 g,用上面方法配對照藥材溶液。按照薄層色譜法試驗[1],在同一塊高效G上點2種溶液,每種4 μL,做條狀,用乙酸乙酯-甲苯-三氯甲烷-甲酸(6:3:5:1)展開,拿出,揮干后噴現(xiàn)配的2%三氯化鐵溶液-1%鐵氰化鉀溶液(1:1),讓斑點顯現(xiàn)清楚。被測顆粒色譜中,白光下顯示和對照品色譜同位置上一樣顏色的斑點,見圖4。
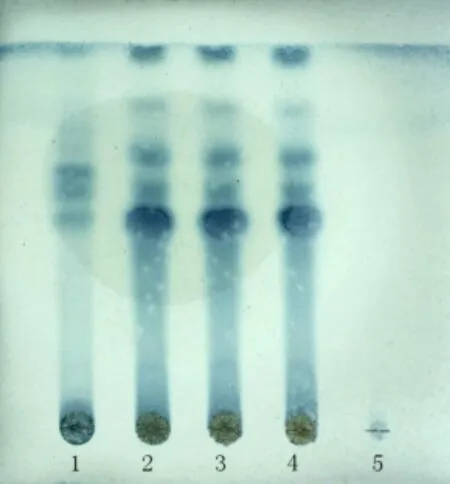
圖4 狗脊 TLC 鑒別
2.1.5 當(dāng)歸鑒別 取QSDH 約15 g,加30 mL 量的水溶解,離心,用乙酸乙酯萃取,取上清液2 次,每次20 mL,歸并乙酸乙酯層,蒸干后加1 mL 甲醇溶成供試品溶液。另取當(dāng)歸對照藥材5 g,加20 mL 量的甲醇,在進(jìn)行30 min 的超聲,濾過,蒸發(fā)濾液,加20 mL 量的水溶解,用乙酸乙酯萃取3 次,10 mL/次,混合乙酸乙酯層,蒸發(fā)后滴1 mL 量的甲醇配成對照藥材溶液。按照薄層色譜法試驗[1],在同一塊高效G上點2種溶液,每種5 μL,用正己烷-乙酸乙酯(4:1)溶液展開,取出,晾干,被測顆粒色譜中,紫外光燈(365 nm)下顯示和對照藥材色譜同位置上一樣顏色的熒光斑點,見圖5。

圖5 當(dāng)歸 TLC 鑒別
2.1.6 陳皮鑒別 取QSDH 約5 g,加20 mL 量的甲醇,超聲處理30 min,取10 mL 量的上清液,濃成1 mL 供試品溶液。另取陳皮對照藥材2 g,用上面方法配對照藥材溶液。再取用甲醇飽和過的橙皮苷對照品作對照品溶液。按照薄層色譜法試驗[1],在同一塊高效G 上點3 種溶液,每種1 μL,用丁酮-乙酸乙酯-甲酸-水(6:10:2:1)溶液展開,取出,晾干,用三氯化鋁試液,被測顆粒色譜中,紫外光燈(365 nm)下顯示和對照藥材色譜同位置上一樣顏色的熒光斑點,見圖 6。

圖6 陳皮 TLC 鑒別
2.1.7 葛根鑒別 取QSDH 約1 g,加入10 mL 甲醇,浸2 h,得到濾液,蒸干后滴0.5 mL 甲醇溶成供試品溶液。另取葛根對照藥材0.8 g,和上面方法配對照藥材溶液。再取葛根素對照品,加甲醇制成1 mL/mg 的溶液,作對照品溶液。按照薄層色譜法試驗[1],在同一塊高效G 上點3 種溶液,每種10 μL,用甲醇-三氯甲烷-水(2.5:7:0.25)溶液展開,拿出,揮干,被測顆粒色譜中,紫外光燈(365 nm)下顯示和對照藥材色譜同位置上一樣顏色的熒光斑點,見圖7。
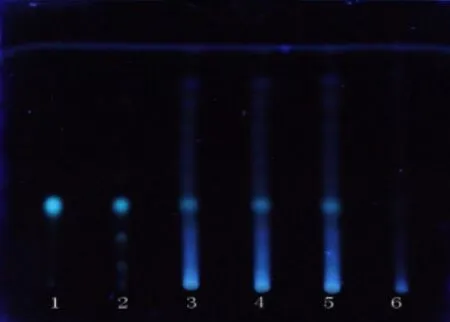
圖7 葛根 TLC 鑒別
2.2 含量測定
2.2.1 色譜條件 色譜柱:島津VP-ODS(4.6×150 mm,5 m);流動相:乙腈-水(32:68 );流 速 :1.0 mL/min;柱溫:30 ℃。檢測器:ELSD,漂移管溫度 :100 ℃,空氣流速:2.8 L/min。理論板數(shù)按黃芪甲苷峰計算應(yīng)不低于4000。
2.2.2 對照品溶液的制備 取黃芪甲苷對照品適量,精準(zhǔn)稱量,混合甲醇配成含0.253 mg/mL 的溶液。
2.2.3 供試品溶液的制備 精準(zhǔn)稱量QSDH 約5 g,研細(xì),混合50 mL 量的甲醇,稱定重量,超聲處理30 min,放涼,稱定重量,加甲醇補上減少的重量,濾過,蒸干,加20 mL 量的水,微熱把殘渣溶解,拿水飽和的正丁醇搖勻提取3 次,每次20 mL,混合正丁醇液,用氨試液充分洗3 回,每回20 mL,丟棄氨試液,正丁醇液蒸干后加甲醇溶解,轉(zhuǎn)至2 mL 量瓶中,加甲醇稀釋至刻度,搖勻,即得。
2.2.4 陰性樣品溶液的制備 按2.2.3 項下方法制成無黃芪的陰性樣品溶液。
2.2.5 專屬性考察 按2.2.1 項下方法測定,得對照品、供試品及陰性對照的色譜圖,見圖8。在這條件下,樣品色譜中待測成分峰前后無其他峰干擾,分離效果比較好。
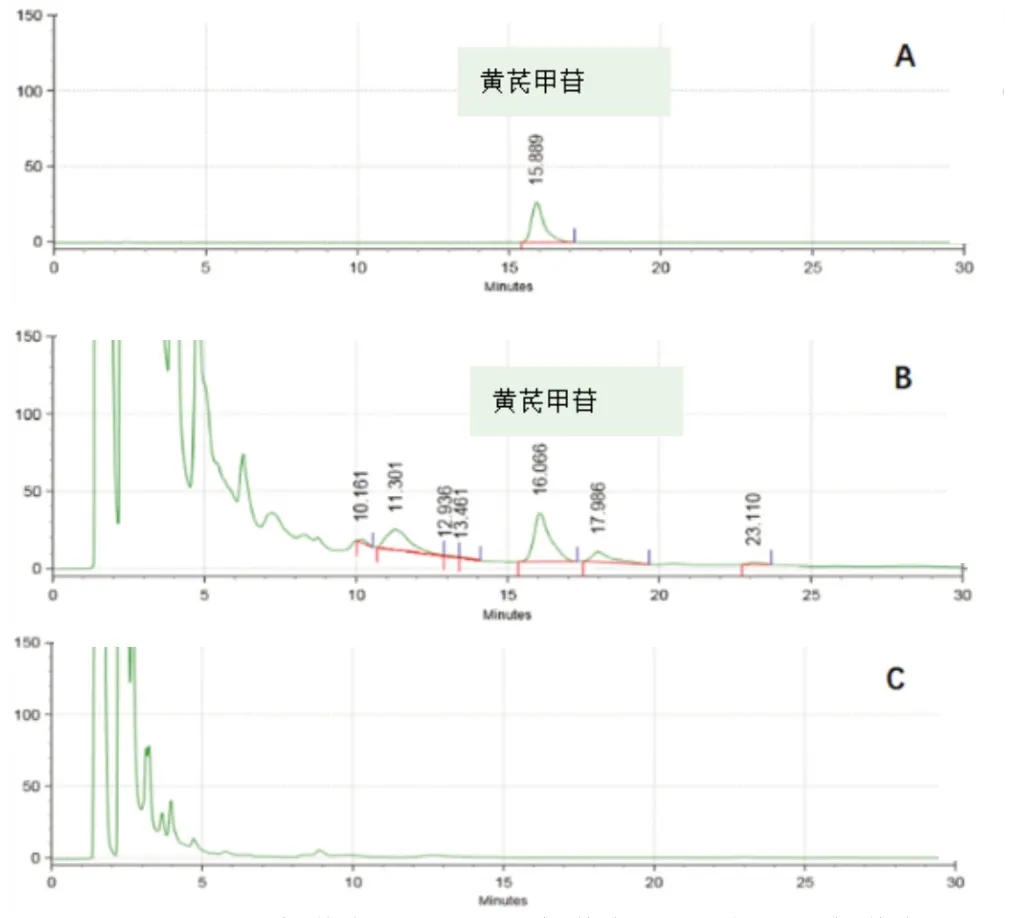
圖8 QSDH 高效液相色譜圖
2.2.6 方法學(xué)考察
2.2.6.1 線性關(guān)系的考察 取2.2.2 項下對照品溶液,分別精密吸取4、8、16、32、64 μL,進(jìn)液相色譜儀,按2.2.1 項下方法測定。水平坐標(biāo)為黃芪甲苷進(jìn)樣量,垂直坐標(biāo)為峰面積的Lg 值繪制標(biāo)準(zhǔn)曲線,得黃芪甲苷的回歸方程為Y(LgA)=1.841X+5.2126,R=0.9997。結(jié)果表明,黃芪甲苷在1.012~16.192 μg 范圍內(nèi)與峰面積的線性關(guān)系比較好。
2.2.6.2 精密度試驗 精密吸取10 μL 2.2.2 項下對照品溶液,按2.2.1 項下色譜條件,連續(xù)測定6 次,結(jié)果黃芪甲苷的RSD為0.25%,證明儀器精密度良好。
2.2.6.3 穩(wěn)定性試驗 精密吸取20 μL 2.2.3 項下中同一份供試品溶液,進(jìn)樣時間0、2、4、6、8、24 h 測定,黃芪甲苷峰面積的RSD為0.30%,證明24 h 內(nèi)供試品溶液基本穩(wěn)定。
2.2.6.4 重復(fù)性試驗 取同一批次芪參地黃顆粒適量,按2.2.3項下方法獨立制備供試品溶液6份并依法測定,黃芪甲苷含量的RSD為0.59%,證明此方法重復(fù)性良好。
2.2.6.5 加樣回收率試驗 取已知含量芪參地黃顆粒6份(黃芪甲苷含量0.0605 mg/g),精密加入黃芪甲苷對照品溶液(0.14 mg/mL)1 mL,依法測定,計算回收率,黃芪甲苷平均回收率為 98.26%,RSD=0.73%。加樣回收率測定結(jié)果,見表1。
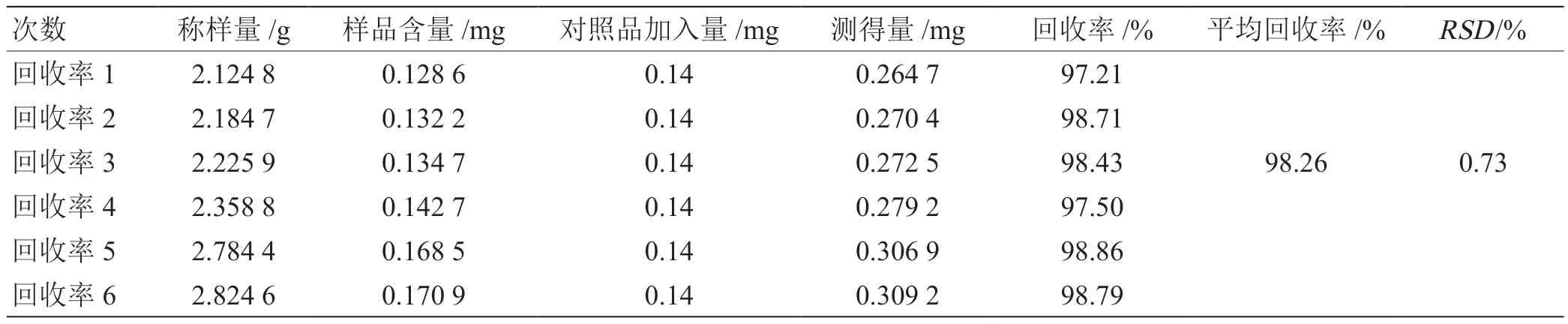
表1 加樣回收率測定結(jié)果
3 討論
黃芪薄層鑒別[2-3]曾用甲醇-三氯甲烷-水(7:13:2)下層溶液展開,但供試品色譜中斑點分離的效果不太好,展開系統(tǒng)極性較低,所以提高了展開系統(tǒng)極性,遂用三氯甲烷-甲醇-水(13:8:2)的下層溶液,且10%硫酸乙醇溶液顯色,斑點不清楚,改成用1%香草醛硫酸乙醇溶液在105℃加熱讓斑點顯色清晰;黨參薄層鑒別[4-5]曾以藥典法中對照品黨參炔苷,展開劑正丁醇-冰醋酸-水(7:1:0.5)10%硫酸乙醇溶液顯色,但供試品色譜中的熒光斑點與對照品對應(yīng)性較差,斑點顯現(xiàn)不清楚,最后改用三氯甲烷-甲醇-水(13:7:2)的下層溶液;當(dāng)歸薄層鑒別[6-7]曾按藥典法乙醚處理對照藥材和樣品,但供試品中無斑點。試用乙酸乙酯進(jìn)行萃取,得到的樣品與對照藥材色譜同位置上,有一樣的熒光斑點,陰性樣品色譜中無一樣斑點;陳皮薄層鑒別[8-9]曾以甲醇-乙酸乙酯-水(17:100:13)為展開劑,展至約3 cm,取出,晾干后用甲苯-乙酸乙酯-甲酸-水(20:10:1:1)的上層溶液為展開劑,展開約8 cm(藥典法),噴顯色劑三氯化鋁試液,放紫外光燈(365 nm)下檢視,但疑似有陰性干擾,因此改用丁酮-乙酸乙酯-甲酸-水(6:10:2:1)為展開劑,噴顯色劑三氯化鋁試液,105℃加熱1 min 后在紫外光燈(365 nm)下檢視;同時枸杞子[10-11]、狗脊[12-14]、葛根[15-17]均經(jīng)過薄層方法學(xué)考察。
黃芪甲苷曾考察了不同的色譜柱、流速、柱溫、流動相,不同的提取方法、溶劑種類、溶劑用量和提取時間,最終確定了準(zhǔn)確可靠,穩(wěn)定可行的方法,為后續(xù)質(zhì)量標(biāo)準(zhǔn)的建立提供基礎(chǔ)。
