美洛昔康納米混懸液的制備及質量評價
2021-10-14 09:50:50李志明呂亞男王琳琳席欣然王愛萍
煙臺大學學報(自然科學與工程版) 2021年4期
李志明,呂亞男,王琳琳,袁 夢,席欣然,王愛萍,
(1.煙臺大學藥學院,煙臺大學新型制劑與生物技術藥物研究山東省高校協同創新中心,分子藥理和藥物評價教育部重點實驗室(煙臺大學),山東 煙臺 264005;2.山東綠葉制藥有限公司長效和靶向制劑國家重點實驗室,山東 煙臺 264003)
美洛昔康(Meloxicam,MLX)是一種高效的非甾體抗炎藥(結構見圖1),可選擇性抑制COX-2(環氧化酶-2)同工酶3,主要用于治療類風濕性關節炎、骨關節炎和術后疼痛[1-2]。美洛昔康與阿片類鎮痛藥物相比,具有較低的胃腸道反應,副作用較小,然而MLX屬于BCS (生物藥劑學分類系統)II類藥物,難溶于水,口服吸收起效慢,限制了其在術后急性疼痛領域的應用[3-5]。
圖1 美洛昔康的結構式
提高藥物溶出速率,實現快速起效的方法有多種,如將藥物制成納米晶體混懸液、前體藥物或鹽、固體分散體、環糊精包合物、脂質傳遞系統、自微乳化體系等[6]。其中納米晶體混懸液無需載體材料,僅需少量穩定劑,可增加難溶性藥物的溶解和吸收,且沒有賦形劑帶來的溶血、過敏等毒性問題,不受包封率及載藥量的制約,能滿足MLX高劑量高濃度快速起效,治療急性術后疼痛的需求[7]。為提高納米晶體混懸劑的穩定性,通常需要加入不同類型的潤濕劑和穩定劑等,以降低表面張力,增加分散介質的黏度、降低微粒的沉降速度,或通過靜電或空間效應阻止粒子聚集或晶型轉變等[8]。納米晶體混懸液的制備方法主要有介質研磨法、高壓均質法及噴霧干燥法、乳化法等[9]。
本研究采用介質研磨法制備美洛昔康納米混懸液(MLX-NS)以提高美洛昔康的溶出速率,通過對潤濕劑和穩定劑(離子穩定劑和空間穩定劑)的種類及用量進行篩選,并對研磨工藝進行優化,以制備既可快速起效又可持續釋放藥物的MLX-NS,為用于急性術后疼痛的MLX-NS的研究開發提供依據。
1 實驗部分
1.1 儀器
DYNO-MILL RL型球磨機(瑞士WEB公司);Hei-TORQUE Precision 400型頂置攪拌器(德國Heidolph公司);1260型高效液相色譜儀(美國Agilent公司);3000型激光粒度儀(英國Mastersizer公司);Zetasizer Ultra型納米粒度電位儀(英國Mastersizer公司);XPE205型電子天平(瑞士Mettler Toledo公司);System 860DL 型溶出儀(美國Logan公司);EM-30型掃描電子顯微鏡(韓國COXEM公司);Sigma 700型全自動表面張力儀(瑞典Biolin公司);DSC822e型差式掃描量熱儀(瑞士Mattler Toledo公司);2.5L型凍干機(美國Labconco公司);D2 PHASER型X射線衍射儀(德國Bruker公司)。
1.2 試藥與試劑
美洛昔康(江蘇飛馬藥業有限公司);吐溫20(國藥集團化學試劑有限公司);吐溫80(國藥集團化學試劑有限公司);聚乙烯比咯烷酮(PVP-K 30,國藥集團化學試劑有限公司);泊洛沙姆188(P 188,德國BASF SE公司);司盤20(Span 20,德國Merck KGaA公司);十二烷基硫酸鈉(SDS,湖南爾康制藥股份有限公司);羧甲基纖維素鈉(CMC-Na,上海麥克林生化科技有限公司);膽酸鈉(上海麥克林生化科技有限公司);脫氧膽酸鈉(上海麥克林生化科技有限公司);乙酸銨(天津市恒興化學試劑制造有限公司);甲醇(色譜純,德國Merck KGaA公司)
1.3 潤濕劑表面張力的測定
采用全自動表面張力儀測定表面張力。取適量MLX原料藥置于料筒中,將其浸入到1%的潤濕劑溶液中進行吸附,天平檢測記錄吸附量隨時間變化的曲線,以正己烷的吸附曲線作為標準計算該樣品在不同潤濕劑溶液中的粉體常數。
1.4 HPLC色譜條件
采用Aglient-C18 (250 mm×4.6 mm,5 μm) 色譜柱,以甲醇溶液-0.2 mol/L乙酸銨水溶液(50∶50)為流動相,進樣量10 μL,柱溫37 ℃,流速0.8 mL/min,檢測波長360 nm。
1.5 納米混懸液的制備
采用介質研磨法制備納米混懸液,具體制備方法如下:稱取處方量的MLX原料藥,在攪拌下加入預先溶解的潤濕劑溶液中,于500 r/min條件下攪拌30 min得初混懸液。將初混懸液轉移至研磨罐中,以設定的研磨參數,研磨一定時間得研磨液。按比例向研磨液中加入穩定劑制得質量分數為1.25%的MLX納米混懸液,將混懸液置于室溫下保存待用。另取少量MLX-NS進行凍干,MLX-NS凍干粉用于晶型、形態的檢測。
1.6 納米混懸液的表征
1.6.1 粒度與Zeta電位 取適量MLX-NS,以蒸餾水為分散介質稀釋至適宜質量分數,采用激光粒度儀測定粒徑、Span值,采用納米粒度電位儀測定Zeta電位,測定溫度為25 ℃。
1.6.2 形態 取適量MLX原料藥粉末及MLX-NS凍干粉均勻分散于附有導電膠的樣品板上,真空鍍金后用掃描電子顯微鏡觀察其形貌。
1.6.3 X射線衍射(XRD) 取適量MLX原料藥粉末、MLX-NS凍干粉、空白輔料凍干粉、MLX與輔料的物理混合粉末,放入玻璃樣品板的凹槽中,置于衍射儀樣品臺進行測試,以5 °/min的掃描速度,2 °~40 °的掃描角度為檢測條件,記錄衍射圖。
1.6.4 體外釋放度 采用透析法測定MLX-NS體外釋放。取MLX-NS 1 mL置于透析袋(14 kU)中,以pH 7.4的PBS 900 mL為溶出介質,控制溶出儀轉速為50 rpm,溫度為37 ℃。分別于溶出5、15、30、60、120、240和480 min 時取樣1 mL,經0.45 μm微孔濾膜過濾,取續濾液得待測樣品,按“1.4”項下的HPLC條件測定并計算MLX的累積釋放量。
1.6.5 穩定性 將制備好的MLX-NS樣品室溫放置,于放置0 d、15 d和30 d時測定粒度及Zeta電位的變化情況,考察制劑的短期穩定性。
2 實驗結果與討論
2.1 潤濕劑潤濕效果的考察
MLX原料藥浸入到質量分數為1%的泊洛沙姆188、吐溫80、吐溫20以及司盤20溶液中的吸附量隨時間變化曲線如圖2所示。不同潤濕劑吸附量隨時間逐漸增加并達到穩定狀態,藥物對潤濕劑的吸附量大小依次為:泊洛沙姆188>吐溫80>吐溫20>司盤20。
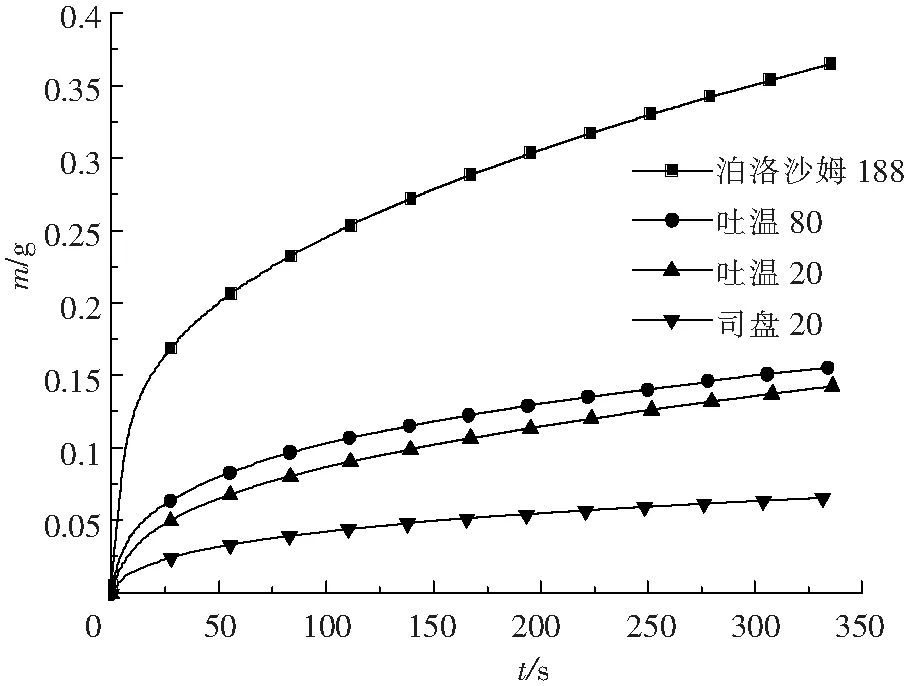
圖2 MLX 在不同潤濕劑中的吸附曲線
通過正己烷得到粉體學常數計算接觸角,藥物與潤濕劑的接觸角大小依次為:吐溫80(89.6)≈吐溫20(89.7)≈司盤20(90.0)>泊洛沙姆188(86.8)。MLX在泊洛沙姆188溶液中的接觸角低于其他三種,潤濕效果最好;MLX在吐溫20、吐溫80及司盤20溶液中的接觸角相近,即潤濕效果接近,然而司盤20在水中難溶,形成具有乳光的非均相體系,不適合單獨作為潤濕劑。因此選擇泊洛沙姆188、吐溫20和吐溫80進一步考察。
分別采用泊洛沙姆188、吐溫20和吐溫80為潤濕劑進行研磨,其粒度結果如表1所示。由表中數據可見泊洛沙姆188和吐溫80作為潤濕劑時粒度分布較寬,Span值大于5,且d90大于1 μm,不符合靜脈注射納米混懸劑的通常要求[10]。分析原因可能是由于泊洛沙姆188和吐溫80溶液更易起泡,在研磨過程中部分粒子殘存在研磨液上層氣泡中致研磨不充分,存在部分大粒子。因此研究選擇吐溫20作為潤濕劑。
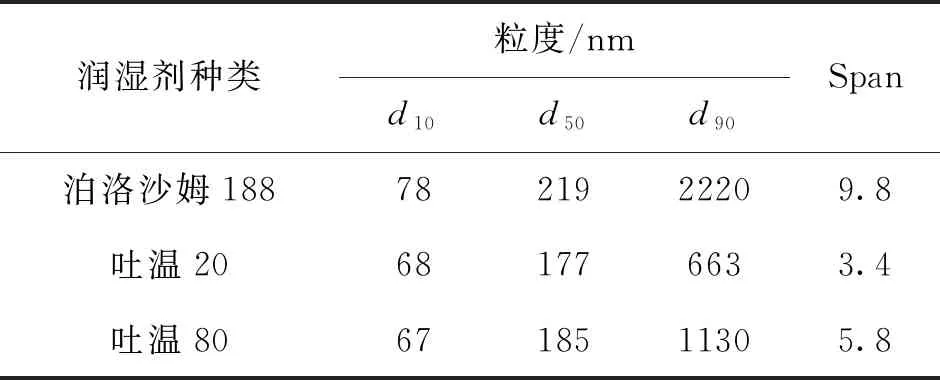
表1 不同潤濕劑研磨的MLX-NS粒度分布
2.2 潤濕劑用量的選擇
以粒度、體外釋放度及短期穩定性數據作為評價指標對潤濕劑吐溫20的用量進行考察,粒度結果如表2所示,三種不同質量分數的吐溫20作為潤濕劑均能降低納米粒子的比表面能,使藥物顆粒研磨至目標粒度。當ω(吐溫20)為0.75%時研磨液的d90顯著高于其他兩個質量分數,且Span值較大,這可能是由于兩親性的吐溫20質量分數過高,使已分散的納米粒子重新包裹發生聚集;吐溫20 質量分數為0.25 %和0.50 %時均能研磨至目標粒度,Span值接近,考慮到吐溫20可對難溶性藥物起到增溶效果,為加速納米混懸液的起效,因此選擇0.5 %吐溫20為作為潤濕劑的最優質量分數。

表2 不同質量分數吐溫20作為潤濕劑研磨的MLX-NS粒度分布
2.3 穩定劑種類的選擇
以粒度、體外釋放及短期穩定性數據作為評價指標,考察了空間穩定劑羧甲基纖維素鈉(CMC-Na)、聚乙烯吡咯烷酮(PVP),離子型穩定劑膽酸鈉、脫氧膽酸鈉以及十二烷基硫酸鈉(SDS)對MLX-NS的影響。由表3可見,以空間型穩定劑PVP和CMC-Na制備的制劑,0 d時制劑粒度無明顯差異,但室溫放置30 d后,以PVP為穩定劑的制劑發生明顯聚集,推測是PVP分子對納米粒子產生吸附作用,較長的聚乙烯鏈中對粒子吸附部分的分子鏈的延伸與未吸附的部分相互吸附導致聚集[11-12];離子型穩定劑中,脫氧膽酸鈉制備制劑室溫放置30 d后也發生明顯聚集,推測是脫氧膽酸鈉自身容易產生聚集行為所致[13-14],而膽酸鈉和SDS放置30 d前后,制劑粒度無明顯變化,其中,SDS帶負電荷通過吸附到粒子表面產生靜電斥力,提高了體系的穩定性。
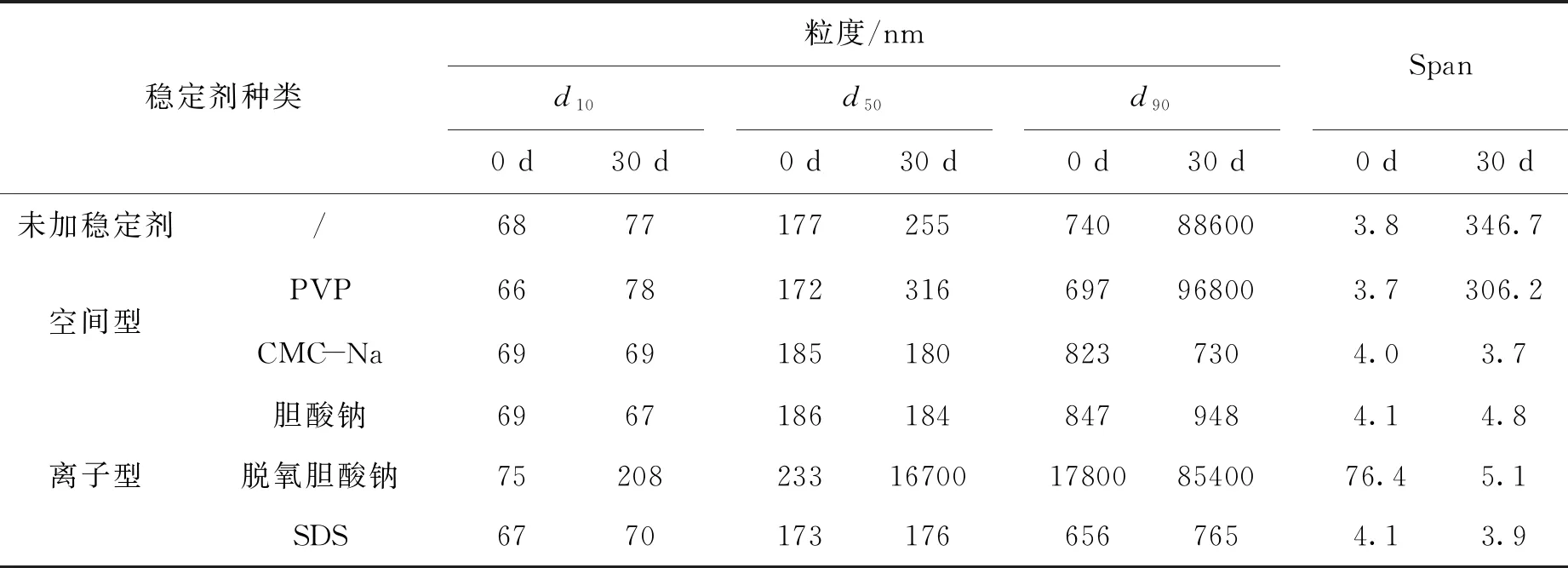
表3 不同穩定劑的MLX-NS粒度分布
將粒度穩定性較好的以CMC-Na、膽酸鈉及SDS為穩定劑制備的制劑進行體外釋放度評價,結果如圖3所示。在相同的研磨液中加入不同的穩定劑,體外釋放曲線明顯不同,不同性質的聚合物穩定劑對納米混懸劑的溶出速率有很大影響[15]。其中以膽酸鈉為穩定劑的MLX-NS釋放最快,2 h內的釋放一直高于其他穩定劑的MLX-NS,且在2 h時釋放完全,這可能是由于膽酸鈉分子具有親水和疏水兩個側面,對美洛昔康有明顯的增溶作用[16],即以膽酸鈉為穩定劑制備的MLX-NS,前期能快速起效,并持續釋放;而以CMC-Na為穩定劑的MLX-NS,前期釋放較慢,后期釋放至8 h仍不足85 %,這可能是由于CMC-Na為親水性穩定劑,對前期美洛昔康的增溶作用無貢獻,且由于CMC-Na分子質量較大,與吸附在納米粒子上的吐溫20親水端形成氫鍵,阻礙了疏水性藥物美洛昔康的溶出擴散[17];以SDS為穩定劑的MLX-NS體外釋放度與未加入穩定劑的MLX-NS無明顯差異,這說明SDS對納米混懸液的快速釋放影響較小,因此為確保MLX-NS達到快速鎮痛的效果,研究選擇膽酸鈉作為穩定劑。
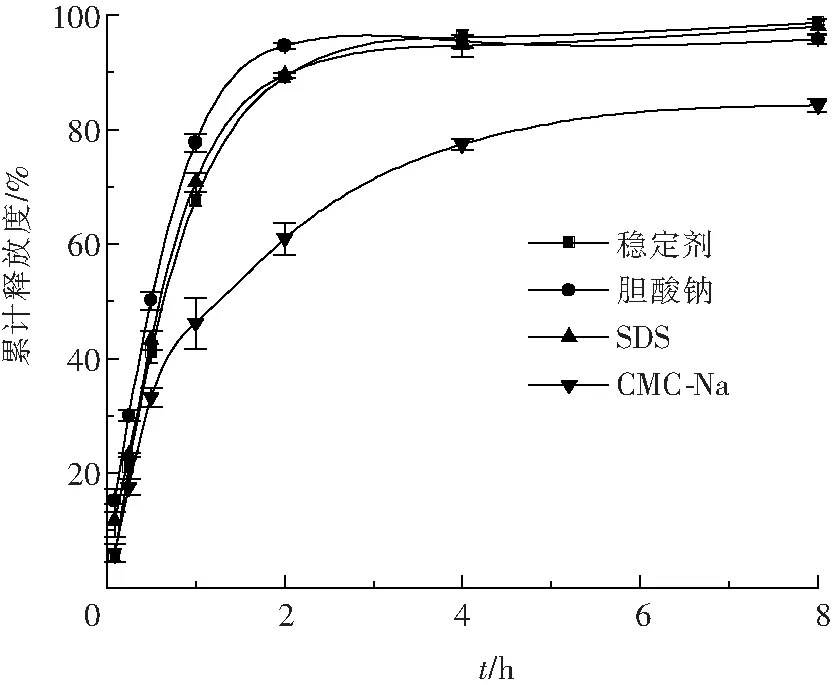
圖3 不同穩定劑的MLX-NS的體外累積釋放曲線(n=3)
2.4 穩定劑用量的選擇
以粒度、Zeta電位為評價指標對膽酸鈉穩定劑的用量進行了篩選,結果如表4所示。加入不同質量分數的膽酸鈉對制劑粒度無明顯影響;電位結果顯示隨著ω(膽酸鈉)的增加,電位絕對值不斷增大,并在0.5 %時趨于穩定,因此選擇0.5 %的膽酸鈉作為穩定劑。有文獻報道,電位絕對值高于20 mV的制劑可以穩定存在[18]。膽酸鈉是陰離子型表面活性劑,常用于制備柔性脂質體,可用于靜脈注射[19],且人體膽汁成分中膽酸鹽含量為7.6 %,因此本研究采用的0.5 %膽酸鈉在人體的安全范圍內[20-21]。
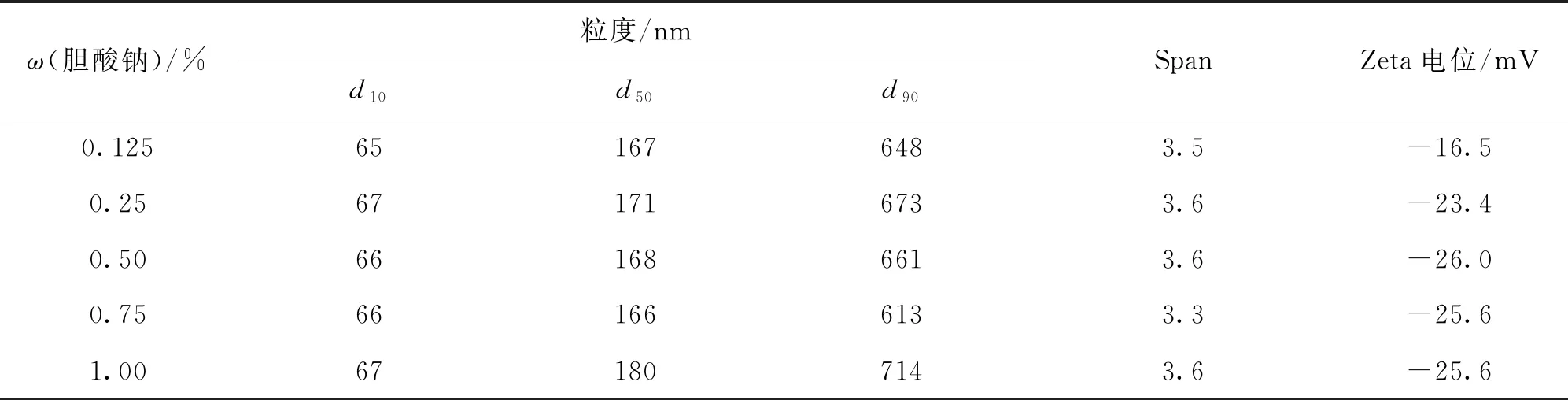
表4 不同質量分數膽酸鈉作為穩定劑的MLX-NS粒度分布及Zeta電位
因此,經過篩選確定最優MLX-NS處方為0.5%吐溫20和0.5%膽酸鈉。
2.5 制備工藝優化
采用最優MLX-NS處方,以粒度分布和研磨效率為評價指標,分別從研磨珠尺寸、研磨轉數以及研磨時間三個參數對制備工藝進行優化。
采用0.8 mm和0.4 mm的研磨珠進行研磨,隨著研磨時間的延長,粒度和Span值均逐漸變小(圖4)。使用0.4 mm研磨珠制備的MLX-NS在研磨相同時間時d50較小、效率較高,且Span值偏小、粒徑分布較均勻,因此選擇0.4 mm的研磨珠進行研磨。
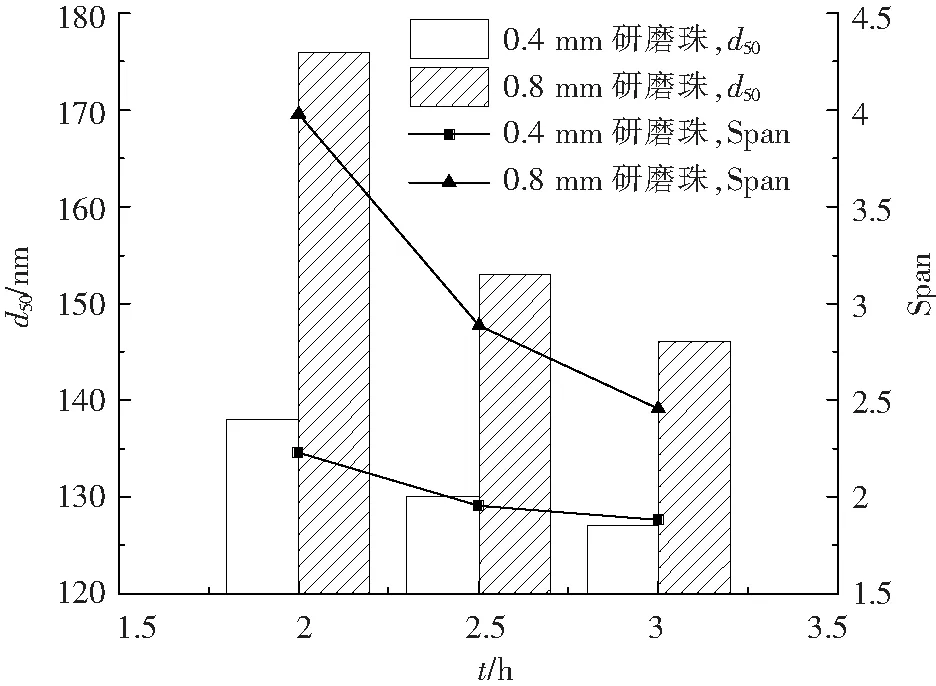
圖4 不同研磨珠尺寸研磨的MLX-NS的粒度分布變化圖
不同研磨轉速下,研磨時間與粒度的關系見圖5。隨著研磨時間的延長,粒度和Span值逐漸降低,并在不同的研磨時間下趨于平衡。由粒度下降趨勢可見,2000 r/min的研磨效率最高,且在2.5 h時粒徑及Span值下降趨勢平緩,因此,選擇研磨轉速2000 r/min,研磨3 h作為最終的工藝參數。
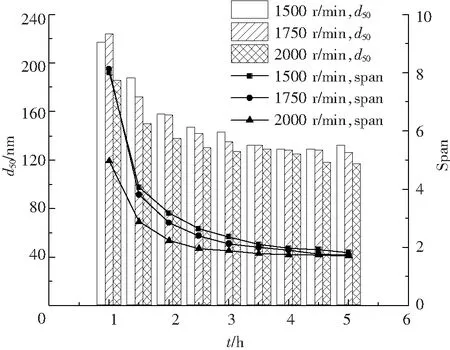
圖5 不同轉速研磨的MLX-NS的粒度分布變化
2.6 MLX-NS制劑的評價
采用最優的處方及工藝制備MLX-NS,并對其粒度、Zeta電位、形態、晶型、體外釋放進行評價。MLX-NS平均粒徑為132 nm,Span值為1.8,粒徑分布較窄,Zeta電位為-26.8 mV。由掃描電鏡圖(圖6)可知MLX原料藥為不規則塊狀晶體,而制成MLX-NS后顆粒尺寸明顯減小,呈現較為均勻的顆粒狀。XRD結果(圖7)顯示研磨前后藥物晶型并未發生改變。體外釋放結果表明(圖8),MLX-NS在5 min累計釋放量達15 %,在2 h時高達92.0%,而原料藥2 h累計釋放量僅為14.6%,這說明將美洛昔康制備成納米混懸劑后可顯著提高藥物的溶出速率。將納米混懸劑在室溫下放置30 d后,粒度及電位無明顯變化(表5),說明穩定性較好,后續將繼續進行穩定性考察。

圖6 MLX和MLX-NS凍干粉的掃描電鏡圖
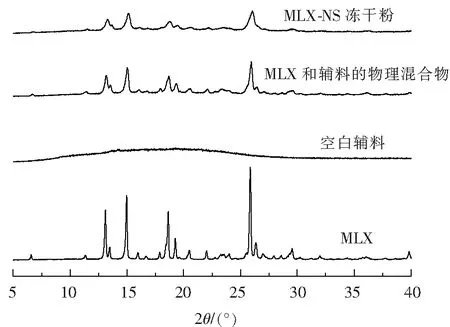
圖7 MLX-NS凍干粉、MLX和輔料的物理混合物、空白輔料、MLX的XRD圖
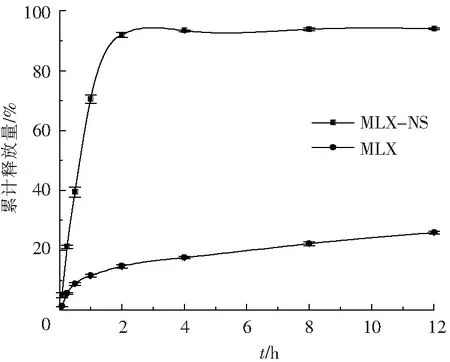
圖8 MLX和MLX-NS體外累積釋放曲線(n=3)
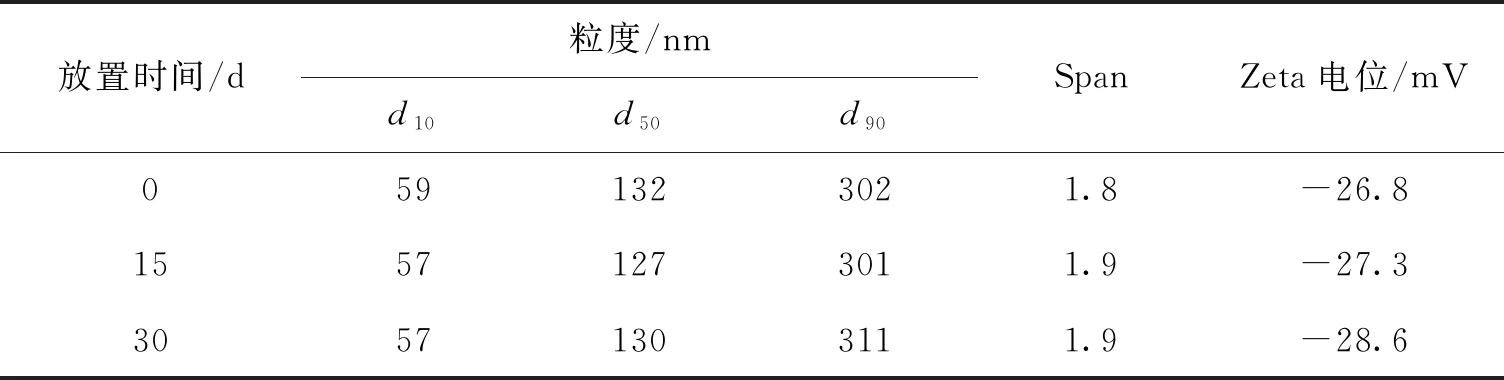
表5 MLX-NS 穩定性結果
3 結 論
采用介質研磨法成功制備了MLX-NS,并對處方和工藝進行了優化。優選的處方以0.5 %吐溫20為潤濕劑,0.5 %膽酸鈉為穩定劑;優化的制備工藝為研磨珠尺寸為0.4 mm,研磨轉速為2000 r/min,研磨時間為3 h。所制備的MLX-NS平均粒徑為132 nm,Zeta電位為-26.8 mV,呈現為較均勻的顆粒狀,研磨前后藥物晶型無顯著變化,MLX-NS在5 min時累計釋放量達15 %,2 h時即可釋放完全,溶出速率顯著提高,常溫放置30 d后制劑性質無顯著性變化,穩定性良好。因此,研究制備的MLX-NS可有效解決美洛昔康難溶性問題,提高美洛昔康的溶出速率,為進一步開發起效快速且可持續釋藥的MLX-NS提供參考。
