面向新一代測序數(shù)據(jù)的病原微生物檢測算法
2021-10-14 06:34:46袁細(xì)國
計(jì)算機(jī)工程與應(yīng)用 2021年19期
李 杰,李 苗,袁細(xì)國
1.西安翻譯學(xué)院 商學(xué)院,西安 710105
2.西安電子科技大學(xué) 計(jì)算機(jī)科學(xué)與技術(shù)學(xué)院,西安 710071
微生物組學(xué)研究是生物信息學(xué)領(lǐng)域的熱點(diǎn)之一,病原微生物在物種水平上的檢測,對于交叉感染疾病和傳染疾病的有效防御和精準(zhǔn)診療具有十分重要的意義和價值。在傳統(tǒng)檢測方法中,往往是通過純培養(yǎng)法培養(yǎng)微生物的方式進(jìn)行觀察和鑒定,但由于可培養(yǎng)的微生物物種極其有限,絕大多數(shù)微生物在現(xiàn)有實(shí)驗(yàn)和技術(shù)條件下難以培養(yǎng),這極大地限制了病原微生物的檢測能力。近些年來,隨著新一代測序技術(shù)(Next Generation Sequencing,NGS)的快速發(fā)展,大批量且分辨率極高的基因組數(shù)據(jù)為微生物的分析提供了新的機(jī)會[1],為從DNA 分子水平上對病原微生物的檢測提供了可靠平臺。從基因組序列角度上看,微生物在物種水平上具有高度相似性,不同微生物物種的差異性堿基占比通常不到全基因組序列堿基數(shù)的百分之五,甚至只有幾十個差異性堿基,這使得微生物物種的鑒別具有較大挑戰(zhàn)性[2]。
微生物組學(xué)的研究對象主要是細(xì)菌,細(xì)菌rRNA按沉降系數(shù)分為3 種:5S、16S 和23S rRNA。16S rDNA(簡稱16S)[3-4]是細(xì)菌染色體上編碼rRNA相對應(yīng)的DNA序列,存在于所有細(xì)菌染色體基因中。從分子水平上分類和鑒定微生物的基本思路是對16S rDNA[2,5]序列進(jìn)行分析,這是因?yàn)?6S rDNA 既具有保守性區(qū)域,又具有高變性區(qū)域[6],可變區(qū)序列因細(xì)菌不同而異,保守區(qū)序列基本保守,使其具備精確刻畫微生物種類和遺傳多樣性的天然條件[7-10]。16S rDNA序列的長度為1 500個堿基左右,大小適中,既能體現(xiàn)不同菌屬之間的差異,又能利用測序技術(shù)較容易地得到其序列。本文均在數(shù)量輕小的16S上進(jìn)行探索,對樣本中物種類型做出鑒定。
早期,基于物種識別的方法使用BLAST(Basic Local Alignment Search Tool)工具[11]將測序讀段局部比對到參考序列,并計(jì)算比對的相似性分?jǐn)?shù)。將測序讀段分配到相似性分?jǐn)?shù)最高的物種下,直至測序讀段全部分配完畢,物種下有測序讀段則樣本中含有該物種。但BLAST 的計(jì)算量較大,所以很多以BLAST 為雛形的算法很難適應(yīng)參考數(shù)據(jù)庫規(guī)模擴(kuò)大或數(shù)據(jù)測序深度增大的情形。另一種物種識別的方法,是基于樣本的共享系統(tǒng)發(fā)育。使用最大似然估計(jì)[12]、貝葉斯后驗(yàn)概率[13]或鄰域連接[14]的系統(tǒng)發(fā)育方法都已經(jīng)開發(fā)出來。用系統(tǒng)發(fā)育的方法來表示生物體之間的關(guān)系是很清晰的,但是這些方法計(jì)算量很大。此外,雖然這些方法能夠進(jìn)行準(zhǔn)確的分類,但系統(tǒng)發(fā)育方法往往靈敏度較低[15]。偽比對是一種基于k-mer[16-18]的快速算法,它使用參考數(shù)據(jù)庫的de Bruijn圖來識別查詢序列的潛在匹配,而不將序列與參考序列對齊。Kallisto[19]是一種基于序列偽比對的物種成分識別算法,通過提取測序讀段間共享的k-mers構(gòu)建de Bruijn 圖,計(jì)算測序讀段來自于某個特定物種的可能性,進(jìn)而判斷樣本中的物種成分。但構(gòu)建de Bruijn圖會占據(jù)巨大的存儲空間。Karp[7]利用了偽對齊的速度和低內(nèi)存要求,并采用EM 算法,該算法使用基本質(zhì)量分?jǐn)?shù)快速準(zhǔn)確地分類微生物組樣本。但是Karp在使用的時候需要輸入很多相關(guān)文件,非專業(yè)人員使用起來相當(dāng)困難。近年來,Lindgreen 等人[20]和Sczyrba等人[21]提出了宏基因組學(xué)中物種檢測方法的性能評估指標(biāo)。
針對傳統(tǒng)培養(yǎng)方法的不足以及現(xiàn)有基于測序數(shù)據(jù)方法的缺點(diǎn),本文提出一種面向新一代測序數(shù)據(jù)的病原微生物檢測算法(NBMicro),將病原微生物樣本測序數(shù)據(jù)比對到參考基因組中,過濾掉比對不成功的測序讀段,針對比對成功的讀段狀態(tài),提取三種有效特征,建立基于樸素貝葉斯分類算法,以對微生物物種存在性進(jìn)行分類(即存在與不存在兩類)。本文采用的樸素貝葉斯分類器,其算法邏輯簡單、分類過程中時間復(fù)雜度和空間復(fù)雜度都較小,且分類效果較好。相比于其他方法,NBMicro 從序列比對結(jié)果中提取的3 種特征,能夠很好地刻畫微生物序列與參考基因組序列的吻合程度,從而有利于判斷微生物物種是否存在。同時,在數(shù)據(jù)量較大的情況下,NBMicro 的計(jì)算時間也較短,操作簡單。
1 研究方法
NBMicro 方法的基本框架如圖1 所示,主要步驟包括數(shù)據(jù)預(yù)處理、特征提取和利用樸素貝葉斯判別物種存在性。首先將數(shù)據(jù)進(jìn)行數(shù)據(jù)預(yù)處理,數(shù)據(jù)預(yù)處理是采用BWA 軟件[22]將待測樣本比對到參考基因組序列(reference,ref)中,將比對不成功的測序讀段(read)去除。其次將預(yù)處理之后的數(shù)據(jù)進(jìn)行特征提取,本文提取物種下比對結(jié)果的3個特征:read depth(讀深)、gapscore、gradmean。read depth 是指ref 中某一片段中比對到的平均位點(diǎn)數(shù);gapscore 是指ref 中未比對到的片段長度和;gradmean是指將ref下比對結(jié)果的read count圖譜的變化特征量化,read count是指ref上每個位點(diǎn)上所比對的read數(shù)量。最后,將提取到的特征使用樸素貝葉斯分類器來判別物種存在性。由于提取到的3 個特征并非在同一標(biāo)準(zhǔn)下,所以先要將3個特征標(biāo)準(zhǔn)化使其在同一標(biāo)準(zhǔn)下,然后利用樸素貝葉斯分類器判別物種是否存在,將存在的物種輸出。
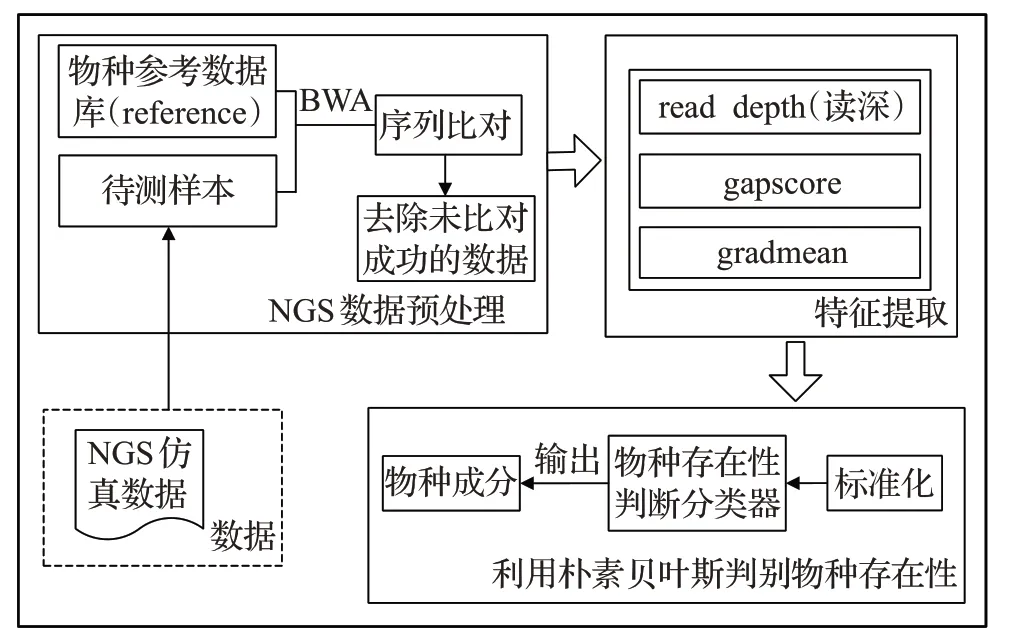
圖1 NBMicro算法流程圖Fig.1 Flow chart of NBMicro algorithm
1.1 特征提取
本文提取了3 種特征:讀深(read depth)、空隙分?jǐn)?shù)(gapscore)、梯度均值(gradmean),其詳細(xì)說明如下所示。
(1)讀深(read depth):是指ref 中某一片段中比對到的read 的位點(diǎn)數(shù)量除以片段長度,read depth 定義如公式(1)所示。樣本組織中如果是含有該物種,那么在測序時該物種的DNA 序列往往會被采集到,從而獲得相應(yīng)的測序讀段。從觀察到的測序數(shù)據(jù)角度上看,若某微生物物種參考基因組比對了一定數(shù)量的讀段(read),就恰好為該物種存在提供了有利證據(jù)。從物種下比對結(jié)果的read depth而言,read depth越高,則該物種比對到的的read越多,該物種存在的可能性越大。

(2)空隙分?jǐn)?shù)(gapscore):是指將ref 下比對結(jié)果的gap 和,其中g(shù)ap 指ref 中未必對到的片段。若物種比對后得到的gap 集合為G={g1,g2,…,gn},gi表示第i個gap 的長度,則物種比對結(jié)果下產(chǎn)生gap 的得分如公式(2)所示。將測序數(shù)據(jù)比對到參考基因組之后,參考基因組中有的位點(diǎn)比對到了讀段,有的沒有比對到,比對到的位點(diǎn)越多,則該物種比對的效果越好。gap 得分將所有未比對到的位點(diǎn)數(shù)統(tǒng)計(jì)起來,可以體現(xiàn)物種比對的效果,從而體現(xiàn)該物種存在的可能性。

如圖2所示是一個ref比對結(jié)果的read count圖譜,圖中一共有7個gap,將g1,g2,…,g7加起來作為物種比對的gap 得分。該物種比對之后發(fā)生的gap 得分越小,說明比對的效果越好,該物種的存在性越大,反之亦然。
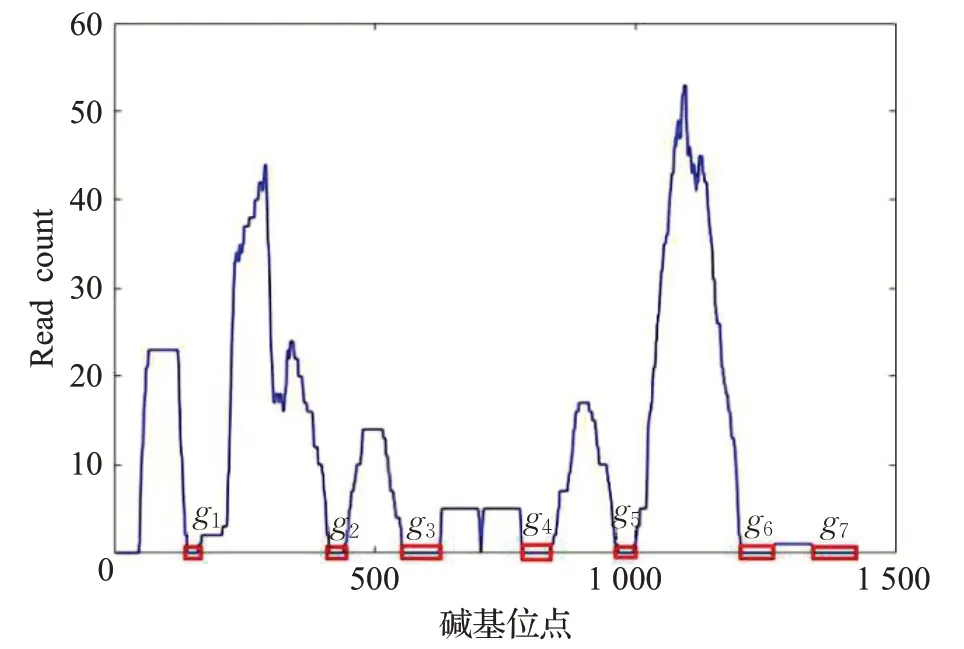
圖2 read count圖譜Fig.2 Read count profile
(3)梯度均值(gradmean):是指將ref中比對結(jié)果的read count 圖譜做傅里葉變換,然后計(jì)算傅里葉頻譜圖上的峰值變化的梯度,并將梯度平均。其中read count是指參考序列上每個位點(diǎn)比對到讀段的數(shù)量,隨著參考序列位點(diǎn)變化的read count可用read count圖譜表示出來;傅里葉變換是指將ref的位點(diǎn)分布函數(shù)變換為ref的頻率分布函數(shù);傅里葉頻譜圖上的峰值變化,即梯度的大小,其意義是ref 上某一點(diǎn)與鄰域點(diǎn)差異的強(qiáng)弱。將測序數(shù)據(jù)比對到參考基因組之后,每一個參考序列都有一個隨位點(diǎn)變化的read count圖譜,將這個read count圖譜進(jìn)行傅里葉變換之后分析隨頻率變化的函數(shù)的峰值,峰值波動小,那么該峰值對應(yīng)的位點(diǎn)與領(lǐng)域位點(diǎn)的差異較小,說明該物種比對效果較好,該物種存在的可能性較大。read count圖譜的傅里葉變換如公式(3)所示:

式中,t表示堿基位置;N表示采樣點(diǎn)數(shù)(即對堿基位點(diǎn)等間隔采樣,采樣最終得到的read count 點(diǎn)數(shù)為N個);u表示頻率,u的取值范圍為,其中fs表示采樣頻率,,T為周期,在這里取2π,u的取值為0,
圖3 顯示了一條ref 隨著位點(diǎn)變化的read count 圖譜,圖4是圖3進(jìn)行傅里葉變換之后的結(jié)果,圖4橫軸表示堿基位點(diǎn)N點(diǎn)等間隔采樣數(shù),其中(N=100)。具體的采樣操作為,將圖3中的1 300個堿基位點(diǎn),每隔13個堿基位點(diǎn)采集一次樣本,一共采集了100 次。圖4 縱軸是信號強(qiáng)度(即read count變換后的信號強(qiáng)度)。圖4中的紅線表示頻譜變化,藍(lán)色點(diǎn)標(biāo)記的是峰值變化。假設(shè)相鄰兩個峰值為P1(v1,w1)和P2(v2,w2),梯度計(jì)算如公式(4)所示:

圖3 ref隨著位點(diǎn)變化的read count圖譜Fig.3 Read count profile of ref as locus changes
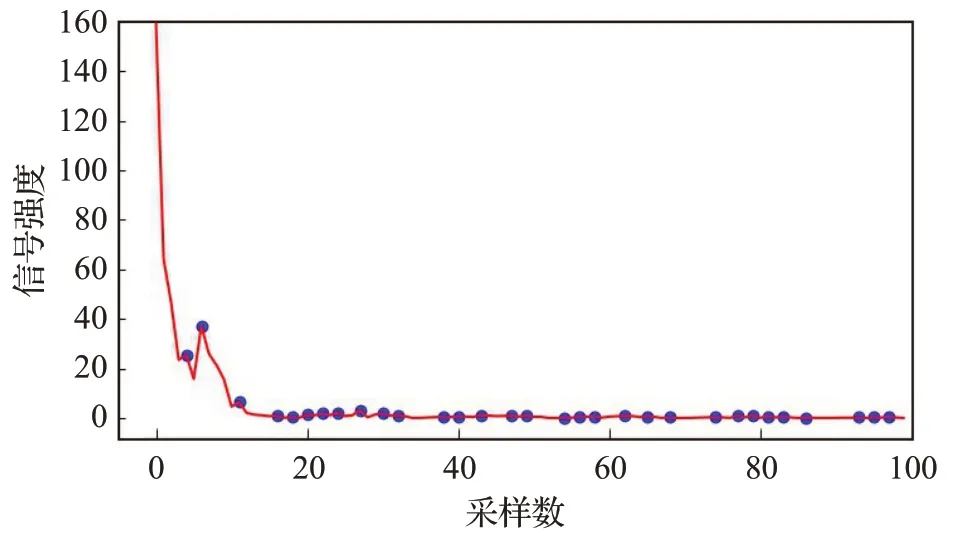
圖4 Read count傅里葉變換圖像Fig.4 Read count Fourier transform graph
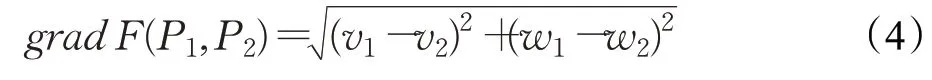
對于每一條ref 求出其隨著位點(diǎn)變化的read count圖譜的傅里葉變換,求出各個相鄰峰值的梯度,再將所有的梯度求均值作為ref 下下比對結(jié)果的一個特征,其公式如(5)所示,其中M表示峰值的數(shù)量。
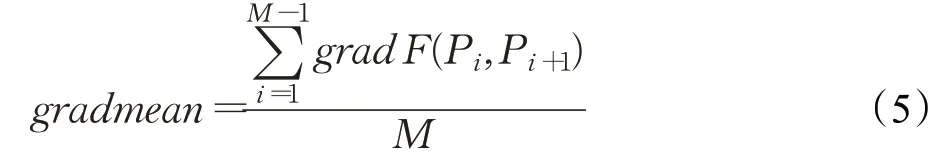
1.2 利用樸素貝葉斯判別物種存在性
基于上述提取的3個特征,采用樸素貝葉斯分類器對物種存在性進(jìn)行判別。樸素貝葉斯是假設(shè)特征之間強(qiáng)獨(dú)立下運(yùn)用貝葉斯定理為基礎(chǔ)的概率分類器。對于給出的待分類項(xiàng),算出在此項(xiàng)出現(xiàn)的特征下各個類別出現(xiàn)的概率,將待分類項(xiàng)分到概率最大的類別下。樸素貝葉斯分類器有算法簡單,并且對小規(guī)模數(shù)據(jù)分類效果好等特點(diǎn)。
微生物物種特征集合X?Rd是d維向量集合,類標(biāo)記集合Y={c1,c2,…,ck},訓(xùn)練數(shù)據(jù)T={(x1,y1),(x2,y2),…,(xD,yD)} 是由P(X,Y) 獨(dú)立同分布產(chǎn)生,其中是第i個樣本的的第j個特征,j=1,2,…,d,yi∈{c1,c2,…,ck}表示第i個樣本分類的取值可能有k個。
類別Y=ck的先驗(yàn)概率如公式(6)所示:
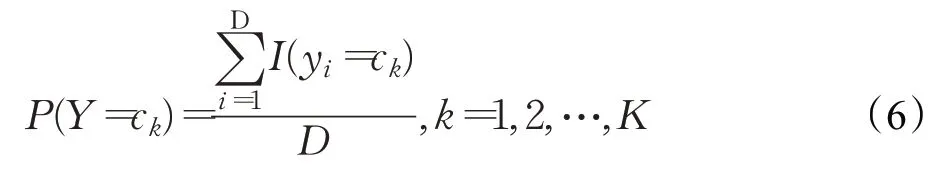
公中,I是指示函數(shù),指微生物物種訓(xùn)練數(shù)據(jù)集合T中哪些類別取值為ck,I(yi=ck)表示如果第i個樣本在類別ck中則為1,否則為0。由于公式(6)會出現(xiàn)概率為0的情況,是指ck中沒有樣本。會影響到后續(xù)的后驗(yàn)概率的計(jì)算,使分類產(chǎn)生偏差,本文采用貝葉斯估計(jì)解決這一問題,其定義公式如(7)所示:
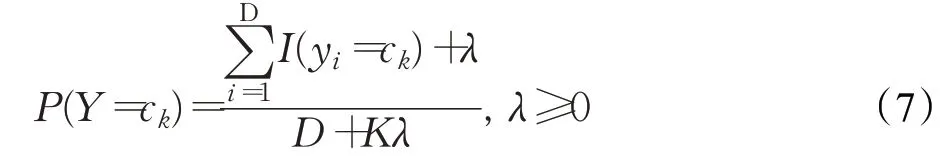
條件概率P(Xj=x|Y=ck)表示Y=ck已發(fā)生的條件下事件Xj=x發(fā)生的概率,由于本文提取微生物物種特征都是連續(xù)分布的,本文使用高斯分布來表示連續(xù)特征的類條件概率分布,其定義如公式(8)所示:
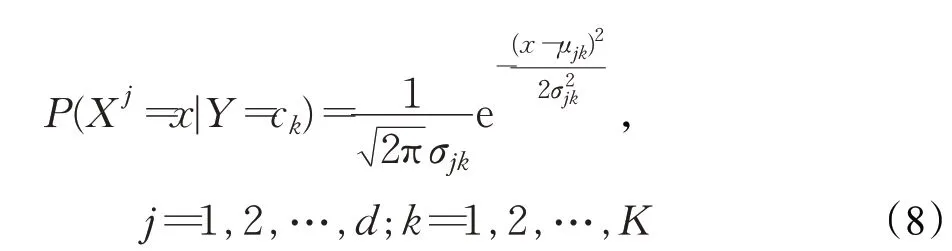
式中,μ表示均值,σ2表示方差,μjk可用類ck的所有訓(xùn)練記錄關(guān)于Xj的樣本均值來估計(jì),可用這些訓(xùn)練記錄的樣本方差來估計(jì)。
樸素貝葉斯法分類時,對于給定的輸入x,通過學(xué)習(xí)到的模型計(jì)算后驗(yàn)概率分布P(Y=ck|X=x),將后驗(yàn)概率最大的類作為x的類輸出,其定義如公式(9)所示:
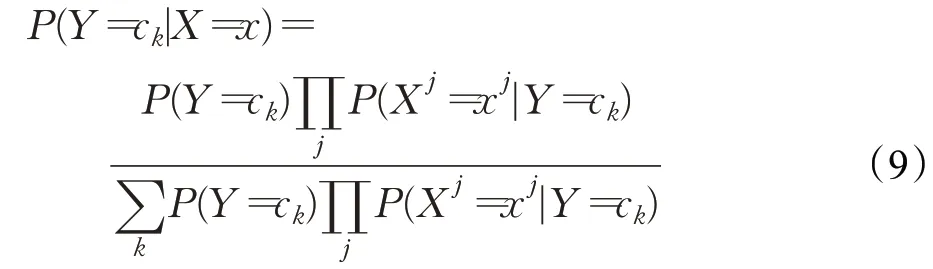
假設(shè)特征條件獨(dú)立,公式(9)中分母對于所有的ck都是相同的,所以可寫為公式(10):
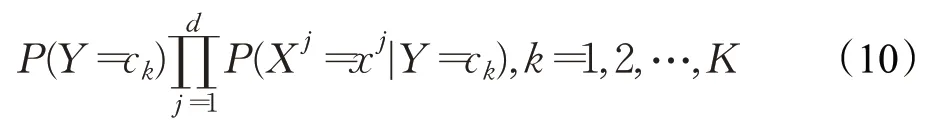
將后驗(yàn)概率最大的類作為x類的輸出,如公式(11)所示:
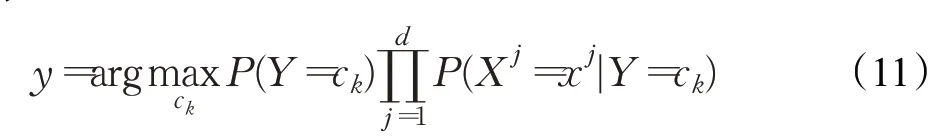
本文1.1 節(jié)提取了每個物種的3 種特征,生成特征向量(read depth、gapscore、gradmean),由于數(shù)據(jù)的不標(biāo)準(zhǔn)會給分類帶來影響,所以這里對特性向量進(jìn)行zscore 標(biāo)準(zhǔn)化。本文的分類只有兩類,若該物種存在則該物種的類別(lable)記作1,否則記作0。對由此得到的數(shù)據(jù)集劃分成8∶2的兩個子樣本,分別用作樸素貝葉斯分類器的訓(xùn)練集和驗(yàn)證集,對物種進(jìn)行存在性判別,并將存在的物種輸出。
2 仿真實(shí)驗(yàn)與分析
仿真實(shí)驗(yàn)對于算法性能的評估十分重要,本文采用ART軟件[23]仿真數(shù)據(jù),設(shè)計(jì)了11組小規(guī)模數(shù)據(jù)和5組較大規(guī)模的數(shù)據(jù)。
為了訓(xùn)練分類器來檢測樣本的中物種的成分,設(shè)計(jì)了11組小規(guī)模不同成分的樣本。由于數(shù)據(jù)庫中物種的相似度過高,匹配到相似物種的可能性較大,這里加入其他未知物種作為干擾。為了與真實(shí)樣本受未知物種干擾的事實(shí)相一致,在仿真樣本中添加了不同干擾程度的未知物種,干擾程度范圍在[0,2.0],以0.2 為步長增長。每組樣本的對應(yīng)數(shù)據(jù)庫有20 條物種,選取其中10條物種和不等量的干擾物種,并使用ART 軟件生成仿真數(shù)據(jù),仿真數(shù)據(jù)的覆蓋度為800X,read長度為75。具體干擾程度如表1所示。
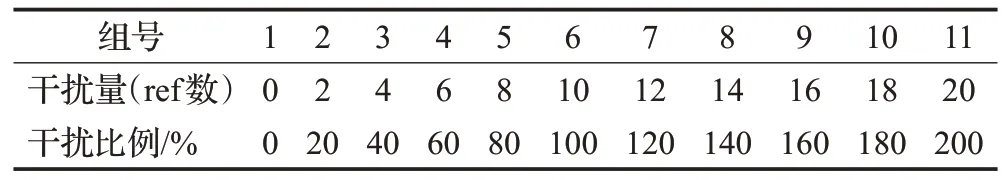
表1 仿真樣本表Table 1 Simulation sample table
本文考慮了實(shí)際情況下人類測序讀物(簡稱人類讀物)的干擾,然后設(shè)計(jì)了5組樣本,其中人類測序讀物與微生物測序讀物(簡稱微生物讀物)的比例分別為0∶10、2∶8、4∶6、6∶4和8∶2。對于每個組,生成6個具有不同程度未知物種干擾的干擾樣本的模擬樣本,這些干擾樣本的不相關(guān)物種干擾程度以步長0.2 在范圍[0,1.0]中變化。每個樣本的生成方法與11組小規(guī)模樣本的相同。
本文選取3種指標(biāo)評價成分檢測的性能,分別是P(精度)、R(召回率)和F1-score 指標(biāo)。F1-score 值越大,成分檢測的性能越好。F1-score指標(biāo)如公式(12):

圖5是在不同干擾程度的樣本上的P值和R值,選取了Karp、Kallisto方法與NBMicro方法做對比,不相關(guān)物種的干擾程度從0 逐步增加到2.0。圖5 橫坐標(biāo)表示不同干擾程度,縱坐標(biāo)表示樣本的P值和R值,其中Karp_R表示Karp 方法的R值,Kallisto_R表示Kallisto方法的R值,NBMicro_R表示本文方法的R值,Karp_P表示Karp 方法的P值,Kallisto_P表示Kallisto 方法的P值,NBMicro_P表示本文方法的P值。從圖5中可以看出,3種方法的R值都很高,說明但凡認(rèn)定物種存在,則幾乎是正確的。3 種方法中NBMicro 的P值較其他兩種高,說明NBMicro將存在的物種識別的效率更高。
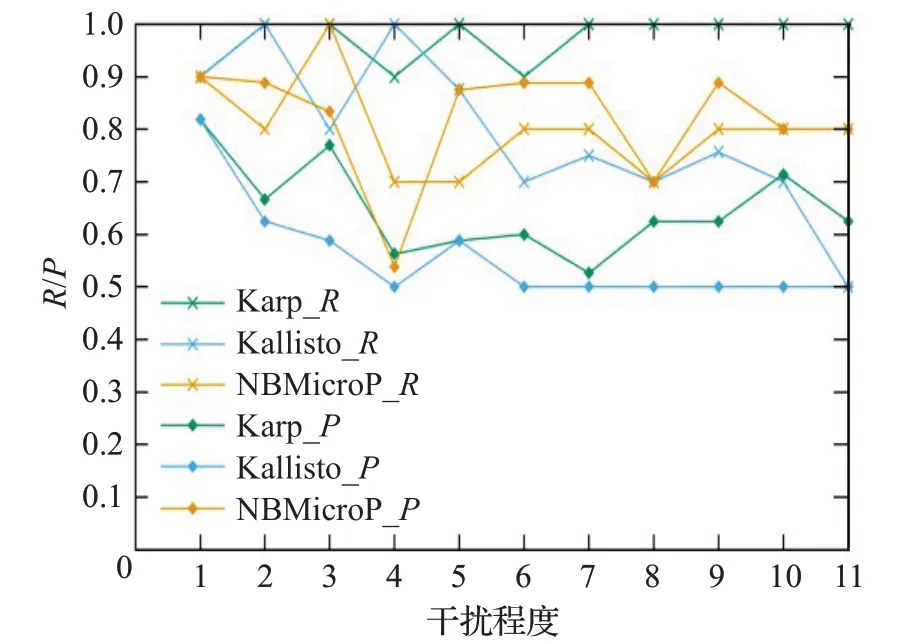
圖5 11組樣本上的R和PFig.5 R and P on 11 samples
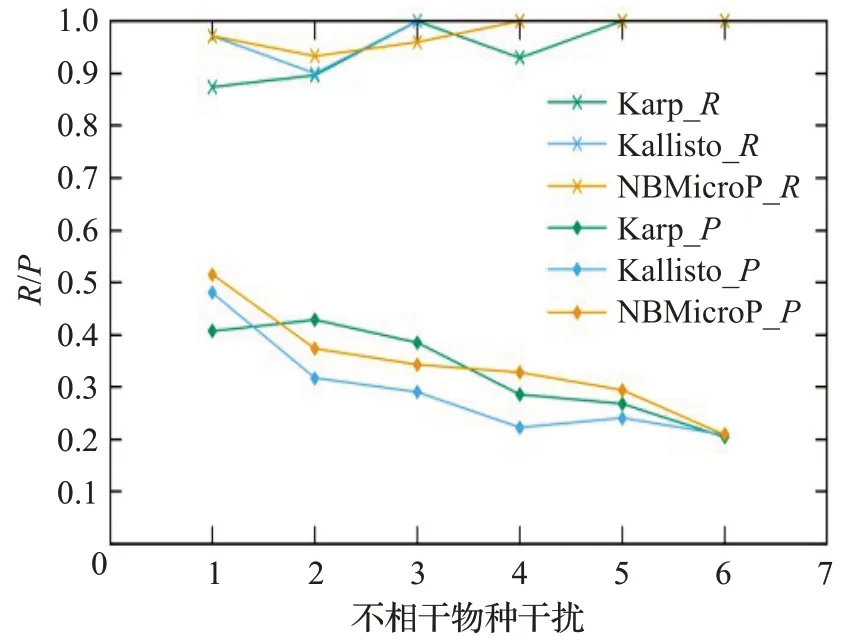
圖6 人類讀物與微生物讀物比例0∶10的R和PFig.6 R and P when the ratio of human read to microbiological read is 0∶10
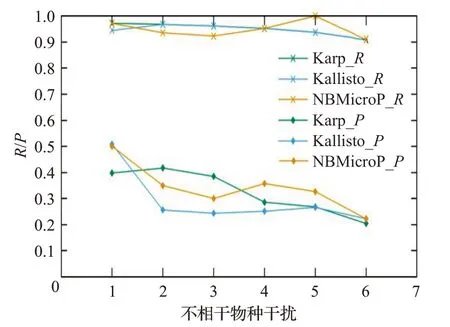
圖7 人類讀物與微生物讀物比例2∶8的R和PFig.7 R and P when the ratio of human read to microbiological read is 2∶8
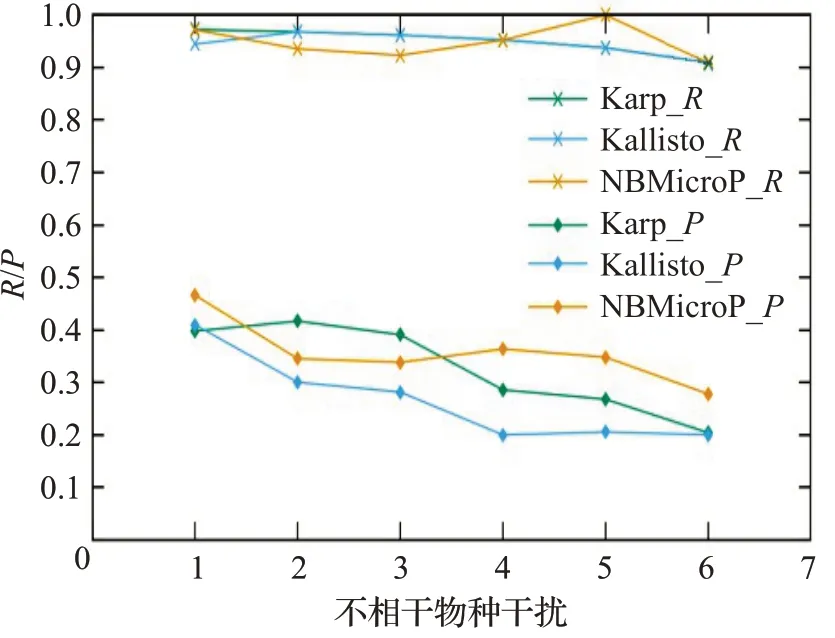
圖8 人類讀物與微生物讀物比例4∶6的R和PFig.8 R and P when the ratio of human read to microbiological read is 4∶6
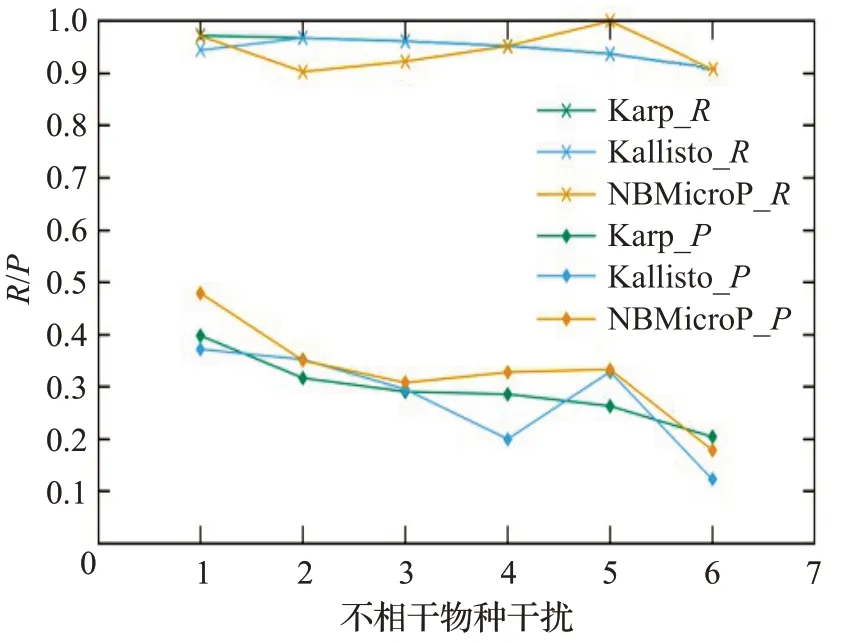
圖9 人類讀物與微生物讀物比例6∶4的R和PFig.9 R and P when the ratio of human read to microbiological read is 6∶4
圖6~10分別對應(yīng)于5組較大規(guī)模樣本的P值和R值,其中人類測序讀數(shù)與微生物測序讀數(shù)的比率分別為0∶10、2∶8、4∶6、6∶4 和8∶2。每個圖都是確定了人類測序讀數(shù)的比率,不相關(guān)物種的干擾程度從0逐步增加到1.0 時(從0.2 逐步增加到0.2)3 種方法的P值和R值。可以清楚地看到,3種方法的R值都較高,接近與1。在P值方面,NBMicro 方法總體上略高于其他兩種方法。隨著不相關(guān)物種的干擾程度的增加,所有方法的P值明顯降低,說明隨著不相干物種的增加,存在的物種的識別效率會有所影響。然而R值不隨干擾程度變化而變化,說明算法識別出的物種的正確率不隨干擾程度變化。此外,圖6~10 的基本走勢相似,說明P值和R值不受人類測序讀物的干擾的影響。
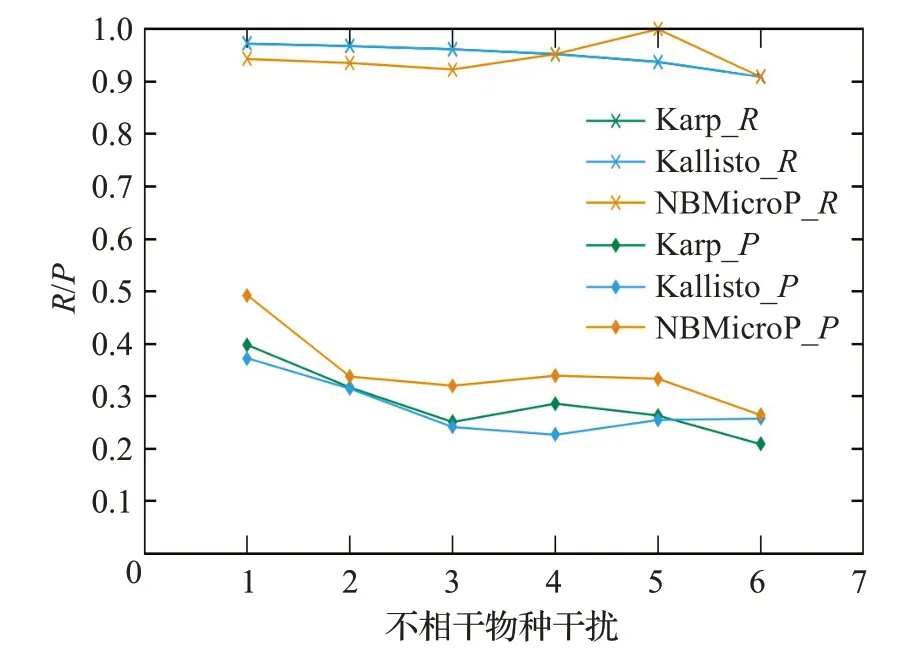
圖10 人類讀物與微生物讀物比例8∶2的R和PFig.10 R and P when the ratio of human read to microbiological read is 8∶2
圖11是在不同干擾程度的樣本上的F1-score值,本文選取了Karp、Kallisto 和Mothur[24]方法與NBMicro 方法做對比,不相關(guān)物種的干擾程度從0逐步增加到2.0。黑色線代表NBMicro 方法的F1-score 值,就整體而言NBMicro方法的物種識別度比其他3種方法高。
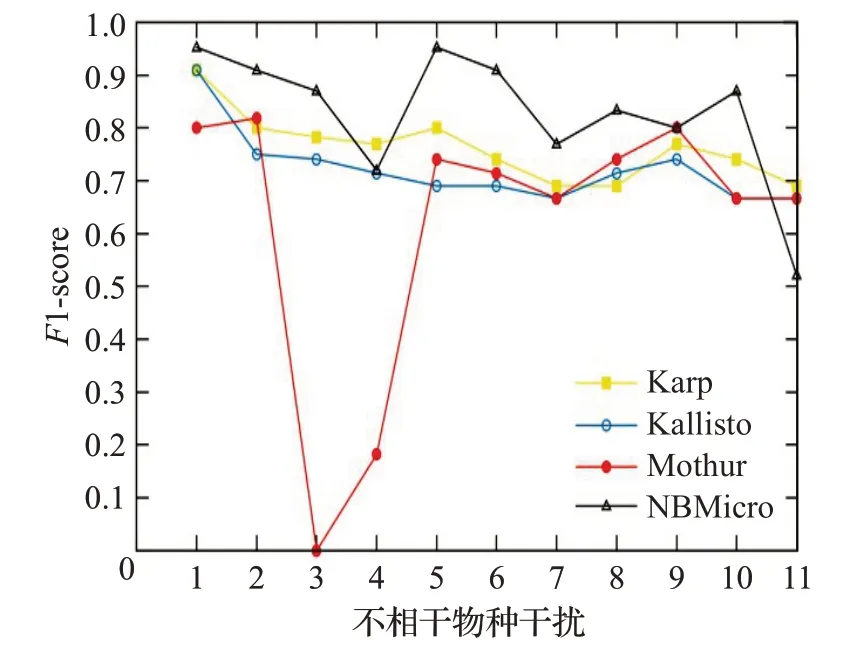
圖11 11組樣本上的F1-scoreFig.11 F1-score on 11 samples
圖12~16 分別對應(yīng)于5 組樣本,其中人類測序讀數(shù)與微生物測序讀數(shù)的比率分別為0∶10、2∶8、4∶6、6∶4和8∶2。每個圖都是確定了人類測序讀數(shù)的比率,不相關(guān)物種的干擾程度從0 逐步增加到1.0 時(從0.2 逐步增加到0.2)4 種方法的F1-score。圖12~16 中黑色的折線代表NBMicro 方法的F1-score 值,可以清楚地看到,NBMicro 方法在物種識別方面較其他方法有較高的準(zhǔn)確性。此外,隨著不相關(guān)物種的干擾程度的增加,所有方法的F1-score明顯降低,說明隨著干擾程度的增加物種識別的準(zhǔn)確度不斷降低,那么物種識別受到了干擾程度的影響。圖12~16的基本走勢不變,說明不受人類測序讀數(shù)影響。
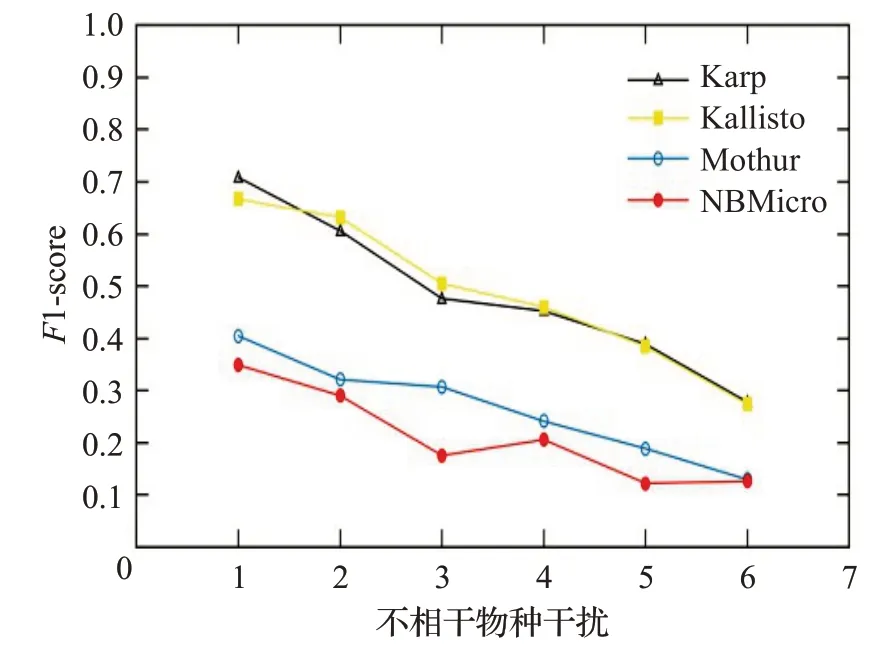
圖12 人類讀物與微生物讀物比例0∶10時的F1-scoreFig.12 F1-score when the ratio of human read to microbiological read is 0∶10
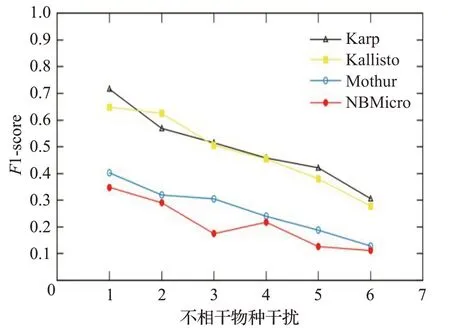
圖13 人類讀物與微生物讀物比例2∶8時的F1-scoreFig.13 F1-score when the ratio of human read to microbiological read is 2∶8
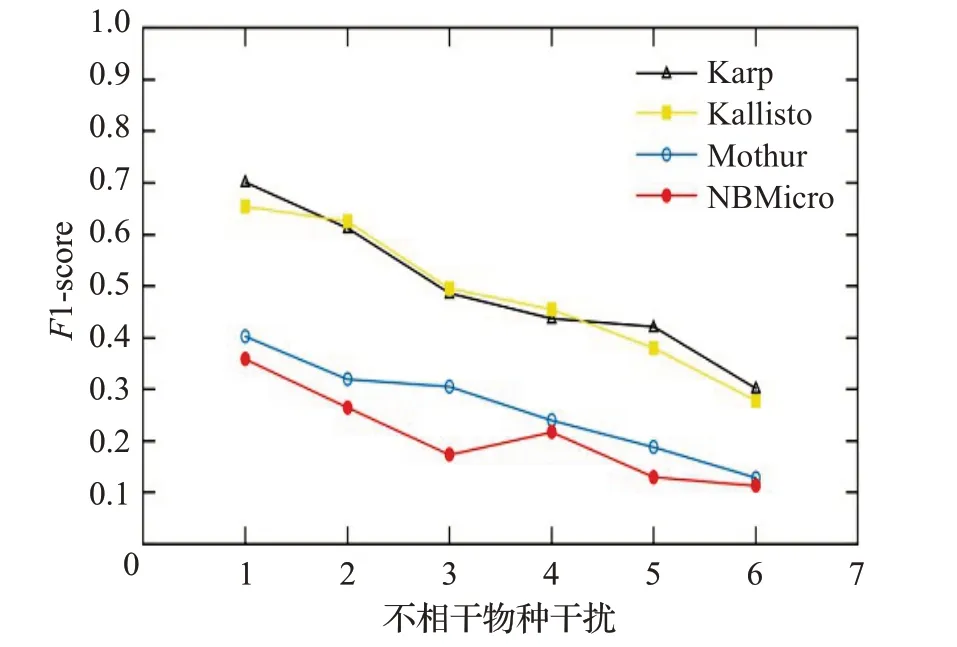
圖14 人類讀物與微生物讀物比例4∶6時的F1-scoreFig.14 F1-score when the ratio of human read to microbiological read is 4∶6
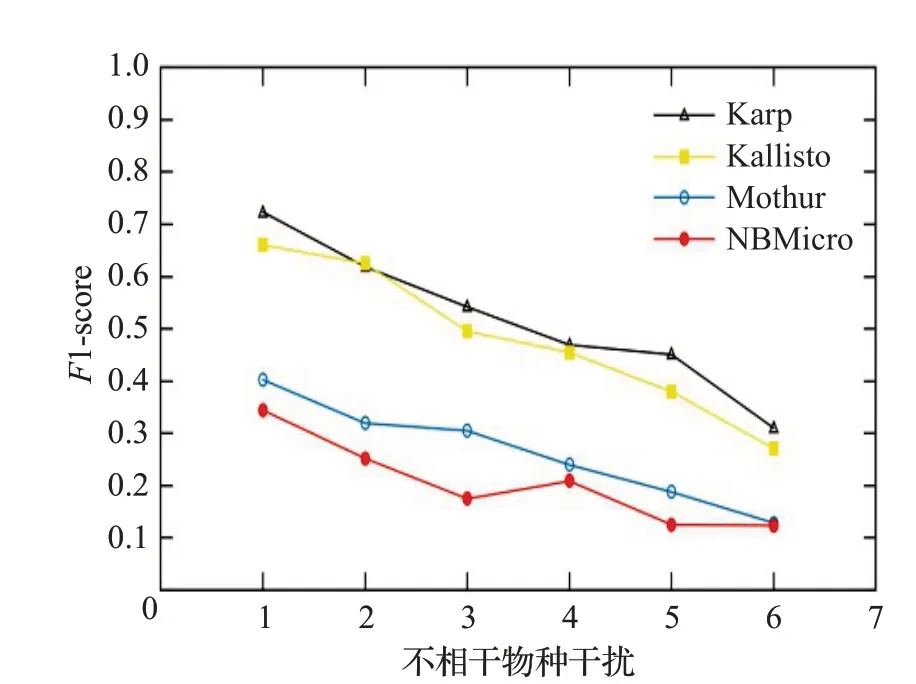
圖15 人類讀物與微生物讀物比例6∶4時的F1-scoreFig.15 F1-score when the ratio of human read to microbiological read is 6∶4
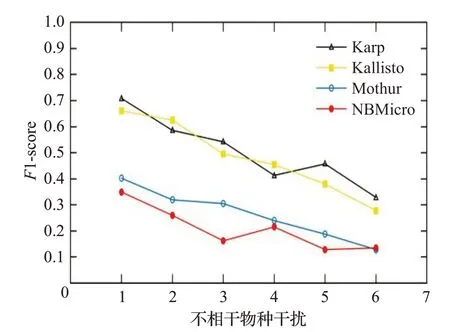
圖16 人類讀物與微生物讀物比例8∶2時的F1-scoreFig.16 F1-score when the ratio of human read to microbiological read is 8∶2
從上述實(shí)驗(yàn)結(jié)果上看,本文方法在不同仿真情景下,都有較好的檢測性能。其最主要的原因在于,所提方法不僅關(guān)注比對讀段數(shù),而且關(guān)注了比對讀段在參考序列上的分布狀態(tài)。由于讀段通常較短,會存在多比對和錯誤比對情況,若僅僅關(guān)注比對讀段數(shù),會引起微生物物種判斷的偏差,那么,比對讀段的分布狀態(tài)能夠提供額外有用信息,這也促使本文方法在數(shù)據(jù)受到干擾情況下也能保持較好檢測性能的原因。
為了驗(yàn)證本文提出的3種特征:read depth(讀深)、gapscore 以及gradmean,對本構(gòu)建的微生物檢測算法的貢獻(xiàn)度或重要性的差異,本文設(shè)計(jì)了一個實(shí)驗(yàn)。選取其中的兩個特征在本文設(shè)計(jì)的11組小規(guī)模數(shù)據(jù)上進(jìn)行檢測,并且計(jì)算檢測結(jié)果的P值、R值和F1-score 值,如果其檢測結(jié)果較差,則意味著沒被選的那個特征比較重要,反之亦然。如圖17 是分別選擇其中兩個特征進(jìn)行檢測的結(jié)果,圖中的“readdepth_R”是指使用了gapscore和gradmean 兩個特征進(jìn)行檢測的結(jié)果的R值,圖中的條形越高說明此特征越不重要。從圖17 中可以看出gapscore 和gradmean 對本文的貢獻(xiàn)較大,而read depth這一特征對本文方法的貢獻(xiàn)度相對較小。
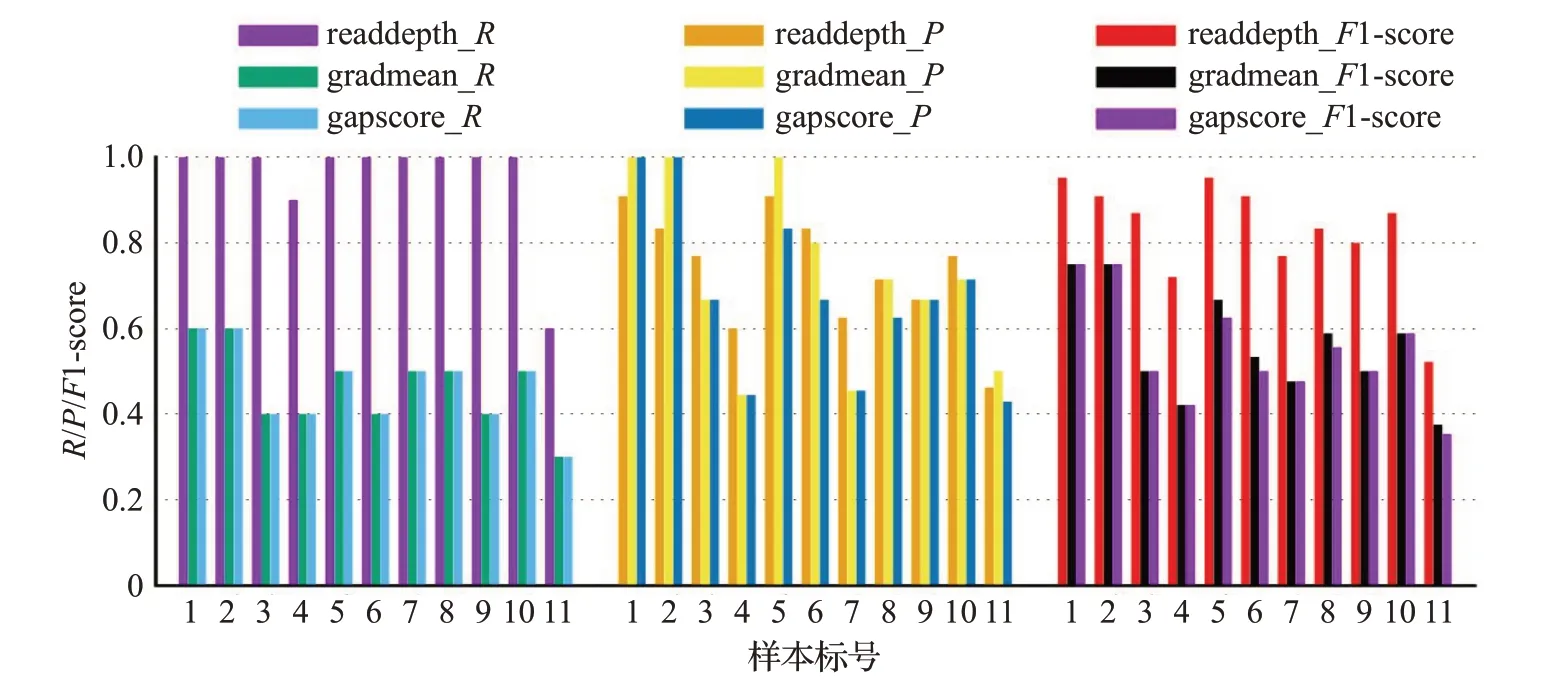
圖17 readdepth、gapscore、gradmean重要程度對比Fig.17 Comparison of importance of readdepth,gapscore and gradmean
3 結(jié)論
本文提出一種面向新一代測序數(shù)據(jù)的病原微生物檢測算法NBMicro,其思路是首先將測序數(shù)據(jù)進(jìn)行預(yù)處理操作后,提取每個微生物物種參考基因組比對結(jié)果的3種特征,然后使用樸素貝葉斯分類器預(yù)測每個物種是否存在。通過仿真數(shù)據(jù)對所提算法進(jìn)行驗(yàn)證,并與同行方法做了比較與分析,表明了所提算法的有效性和優(yōu)勢。因此,本文算法NBMicro 對于從DNA 分子數(shù)據(jù)上分析微生物物種具有推動作用。另外,本文還存在不足之處,比如微生物種群數(shù)據(jù)庫規(guī)模有限,且僅考慮了微生物物種的檢測,卻沒有考慮樣本中每種微生物物種含量的估計(jì),這在今后工作中將進(jìn)一步完善。
猜你喜歡
課堂內(nèi)外·初中版(科學(xué)少年)(2025年1期)2025-02-28 00:00:00
課堂內(nèi)外·初中版(科學(xué)少年)(2025年2期)2025-02-28 00:00:00
英語世界(2023年10期)2023-11-17 09:18:18
兒童故事畫報(bào)(2019年5期)2019-05-26 14:26:14
科學(xué)大眾(中學(xué))(2019年3期)2019-05-17 10:04:30
汽車觀察(2018年10期)2018-11-06 07:05:26
Coco薇(2016年2期)2016-03-22 02:42:52
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年7期)2015-08-11 15:03:12
少兒科學(xué)周刊·少年版(2015年1期)2015-07-07 17:15:12
