高效液相色譜-串聯質譜法檢測雞蛋中的氟喹諾酮類藥物殘留
2021-10-21 06:18:34穆小婷胡克科別又才陳智標
食品工程 2021年3期
關鍵詞:檢測
穆小婷 宋 佳 胡克科 別又才 陳智標
1(清遠市農業科技推廣服務中心,廣東清遠 511500)
2(清遠市農產品質量檢驗檢測中心,廣東清遠 511500)
氟喹諾酮類(FQs)抗生素是人工合成的新型殺菌廣譜型抗菌藥物,通過抑制細菌DNA 螺旋酶,阻斷DNA 復制而產生抗菌作用,對G+、G-、支原體、衣原體均有很好的殺菌作用,因其高效、低毒、價格低廉等特點而被廣泛用于動物和水產養殖中,尤其多應用于治療家禽的感染性疾病。但過量使用氟喹諾酮類抗生素不僅會使生物體產生耐藥性,藥物殘留于畜禽體內,而且,被食用后會累積在人體中,對成人體內各大系統及中樞神經造成不同程度的損傷,長時間食用甚至會致癌、致畸。為了有效地對動物源性食品中氟喹諾酮類藥物殘留進行檢測和監控,國內外建立了多種氟喹諾酮類藥物殘留的檢測方法,常用的檢測方法有高效液相色譜法和液相色譜-質譜法,在頒布的相關標準中,農業部781 號公告-6-2006《雞蛋中氟喹諾酮類藥物殘留量的測定高效液相色譜法》能夠用于檢測雞蛋中氟喹諾酮類藥物殘留量,但缺點是回收率太低,無法滿足當前要求;GB/T 21312—2007《動物源性食品中14 種喹諾酮藥物殘留檢測方法液相色譜-質譜/質譜法》可同時檢測雞肉和雞蛋中的喹諾酮藥物殘留,但在實際操作中提取效果并不理想,達不到檢測分析所需要求。
本文在GB/T 21312—2007《動物源性食品中14 種喹諾酮藥物殘留檢測方法液相色譜-質譜/質譜法》 的基礎上,對標準中的提取步驟進行了優化,并試驗了常用的8 種喹諾酮類藥物的檢測效果,以期為雞蛋中氟喹諾酮類殘留的測定提供參考。
1 材料和方法
1.1 主要試劑和儀器
檸檬酸、碳酸氫二鈉、乙二胺四乙酸二鈉、鹽酸、氫氧化鈉、乙酸銨,均為分析純;甲醇、乙腈,均為色譜純;甲酸,優級純。
HLB 固相萃取柱、電子天平(120 g/0.1mg)、漩渦混合器、高速離心機、超純水機、超聲波清洗器、固相萃取裝置。
1.2 儀器條件
離子源:電噴霧離子源(正離子掃描模式);掃描方式:多反應監測模式;霧化氣:氮氣;碰撞氣:氬氣;毛細管電壓:0.35 kv;源溫度:150 ℃;脫溶劑氣溫度:350 ℃;脫溶劑氣流:1 000 L/Hr;錐孔氣流:30 L/Hr,駐留時間:0.028 s。其他參數如表1 所示。
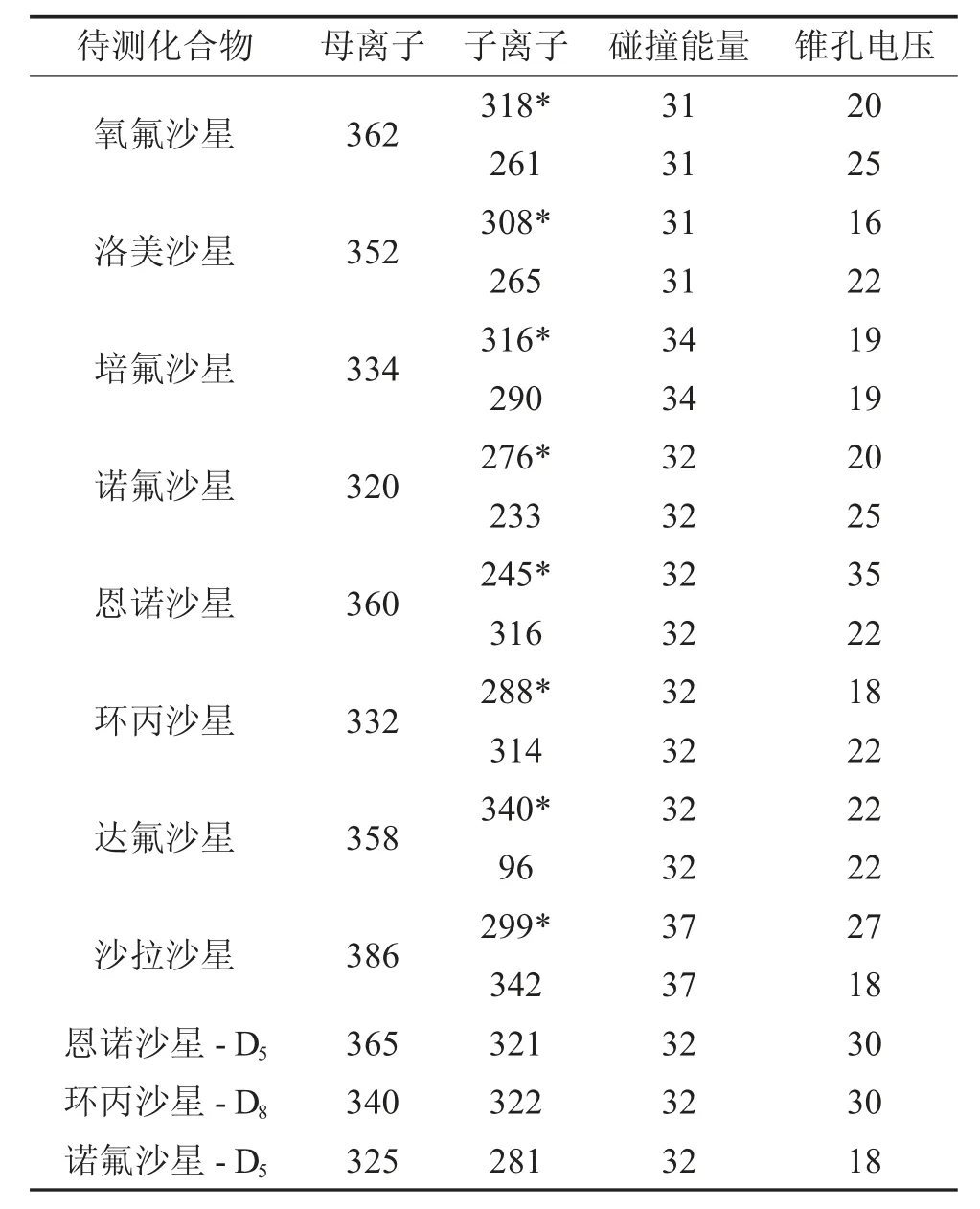
表1 8 種待測物及3 種內標物的監測離子對參數
1.3 樣品前處理步驟
1.3.1 提取
稱取均質試樣5.00 g,置于50 mL 聚丙烯離心管中,加入40 mL 0.1 moL/L EDTA-Mcllvaine 緩沖溶液,1 000 r/min 漩渦混合1 min,超聲提取10 min,-18 ℃冷凍30 min,12 000 r/min 離心10 min(4 ℃),取上清液到另一50 mL 離心管,用0.1 mol/LEDTA-Mcllvaine 緩沖溶液定容到50 mL。吸取20 mL已定容好的提取液到另一50 mL 離心管,加入20 mL正己烷,振蕩1 min,12 000 r/min 離心10 min,取下層提取液到50 mL 離心管。
1.3.2 凈化
先用6 mL 甲醇洗滌、6 mL 水活化HLB 固相萃取柱,后將提取液以2 mL/min~3 mL/min 的速度過柱。棄去濾液,用2 mL 5%甲醇水溶液淋洗,棄去淋洗液。將小柱抽干,再用6 mL 甲醇洗脫并收集洗脫液。洗脫液用氮氣吹干,1 mL 0.2%甲酸水溶液溶解,1 000 r/min 漩渦混合1 min,過孔徑0.22 um濾膜,供高效液相色譜質譜聯用儀測定。
2 結果分析及討論
2.1 方法的優化
2.1.1 提取液pH 的優化
按GB/T 21312—2007 步驟用pH4.0 的Mcllvaine緩沖溶液進行提取時發現,雞肉和雞蛋的提取效果非常差,尤其雞蛋,提取離心后幾乎沒有分層,很難進行下一步試驗。根據氟喹諾酮類的化學結構可知,多數氟喹諾酮類在C-3 處含有羧基,C-7 處連有哌嗪等含氮堿性基團,屬于酸堿兩性化合物,其酸解離常數pKa=5.5~6.6,堿解離常數pKa=7.2~8.9,等電點pKI≈7。鑒于此,本試驗將Mcllvaine 緩沖溶液的pH 調節為7.0,試驗結果顯示,提取后出現明顯的分層,因此本試驗將Mcllvaine 緩沖溶液的pH 調節為7.0。
2.1.2 添加正己烷除脂
GB/T 21312—2007 中離心結束后直接取上清液過柱,可實際試驗發現,因離心時分層效果并不理想,尤其雞蛋的油脂含量高,很難取到上清液,雖然經過提取液pH 的改良,分層效果好很多,但除脂效果還是不理想。因此,考慮加入正己烷除脂。試驗采用上清液和正己烷1∶1 除脂,分取20 mL上清液,結果發現離心后上清液很清澈,回收率也良好,與楊嘉偉試驗結果有所不同,而與楊朝琳的試驗結果相統一。考慮到分取體積太少增大誤差,本試驗最終確定分取體積20 mL,加入正己烷20 mL 進行除脂。
2.1.3 雞肉雞蛋合并提取
GB/T 21312—2007 中雞肉和雞蛋的提取液是同一種,但提取次數及提取液體積不同。雞肉提取步驟為加入20 mL 0.1 moL/L EDTA-Mcllvaine 緩沖溶液,提取3 次,合并上清液。雞蛋提取步驟為加入40 mL 0.1 moL/L EDTA-Mcllvaine 緩沖溶液提取1 次。在實際試驗過程中,大部分情況都是批量試驗,且雞肉雞蛋同一批次做,因此,本試驗研究如何將二者的提取步驟統一。經反復試驗發現,雞肉雞蛋均加入 40 mL 0.1 moL/L EDTA-Mcllvaine 緩沖液提取,提取后將提取液轉入另一離心管后用0.1 moL/L EDTA-Mcllvaine 緩沖液定容至50 mL,這樣提取其結果回收率并不低于原步驟,且比原步驟簡單易操作。
本試驗在離心前增加冷凍處理30 min 的步驟。因少部分樣品脂肪含量太高,經過前面的優化還是不能很好的分層,因此本試驗對冷凍處理和熱處理進行對比,即將樣品放在冰箱中冷凍30 min,或在水浴鍋中進行熱沉淀,之后再進行離心提取,結果發現,分層效果不好的樣品經過冷凍30 min 處理即可得到解決。在實際操作中,若經過前面處理步驟可以得到很好的上清液則不需要進行此步驟。另外,試驗采用性質相似的同位素內標法進行測量,有效降低了前處理過程中的操作誤差和其他條件導致的不統一及基質效應對結果的影響。圖1~圖11為雞蛋中待測物及內標物的定性定量離子色譜圖。
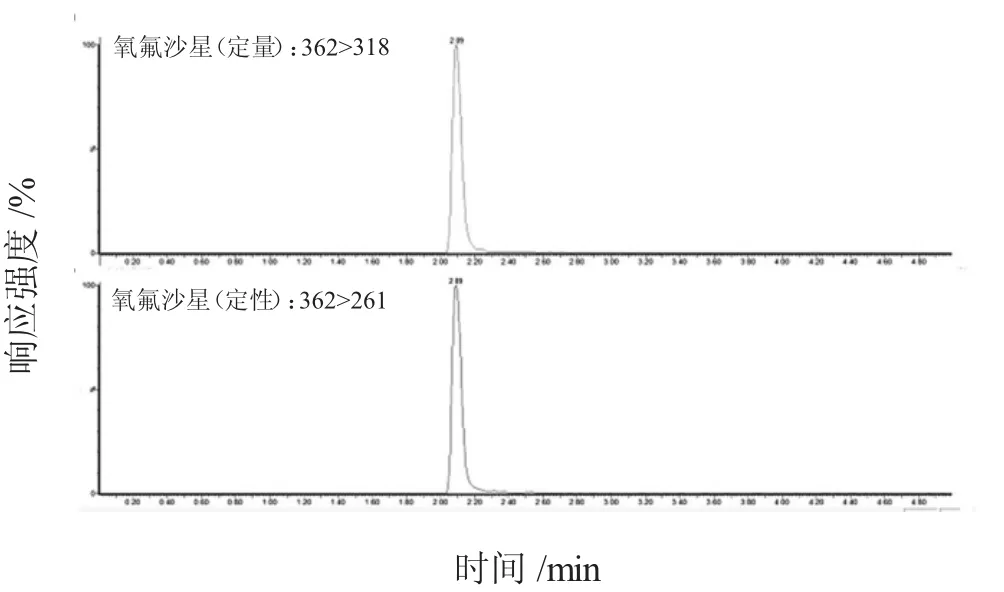
圖1 氧氟沙星定性定量離子色譜圖
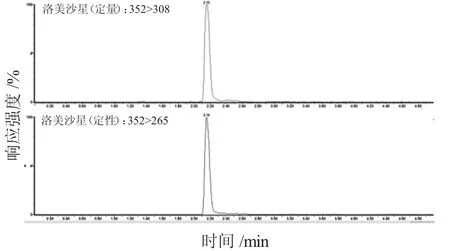
圖2 洛美沙星定性定量離子色譜圖
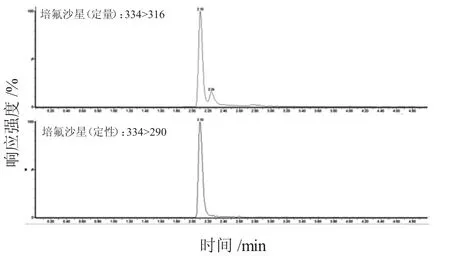
圖3 培氟沙星定性定量離子色譜圖
圖4 諾氟沙星定性定量離子色譜圖
圖5 恩諾沙星定性定量離子色譜圖
圖6 環丙沙星定性定量離子色譜圖
圖7 達氟沙星定性定量離子色譜圖
圖8 沙拉沙星定性定量離子色譜圖
圖9 恩諾沙星-D5 離子色譜圖
圖10 環丙沙星-D8 離子色譜圖
圖11 諾氟沙星-D5 離子色譜圖
2.2 方法學論證
2.2.1 校準曲線和線性范圍
將混合標準工作液用初始流動相(甲醇-乙腈溶液:0.2%甲酸水溶液=1∶9)逐級稀釋成5.0 ng/mL~100.0 ng/mL 的標準系列溶液。平行稱取雞蛋陰性樣品5.00 g,分別置于6 支50 mL 聚丙烯離心管中,加入標準系列溶液1.0 mL,最終形成5.0 ng/mL(1.0 μg/kg)、10.0 ng/mL(2.0 μg/kg)、25.0 ng/mL(5.0 μg/kg)、50.0 ng/mL(10.0 μg/kg)、75.0 ng/mL(15.0 μg/kg)、100.0 ng/mL(20.0 μg/kg)6 個標準系列溶液,按以上優化后的步驟進行提取和凈化。以所得的8 種氟喹諾酮類藥物的離子峰面積為縱坐標,標準溶液的質量濃度為橫坐標,繪制標準曲線。表2 為回歸方程和相關系數。
2.2.2 檢出限和定量限
雞蛋中氧氟沙星、洛美沙星、培氟沙星、諾氟沙星、恩諾沙星、環丙沙星、達氟沙星、沙拉沙星8 種檢測指標在GB/T 21312—2007 中檢出限分別為0.5 μg/kg、0.5 μg/kg、1.0 μg/kg、1.0 μg/kg、0.5 μg/kg、1.2 μg/kg、0.5 μg/kg、1.0 μg/kg,定量限分別為2 μg/kg、2 μg/kg、3 μg/kg、3 μg/kg、2 μg/kg、4 μg/kg、2 μg/kg、3 μg/kg。本試驗以標準曲線上的最低點,取信噪比為S/N=3,結合稀釋倍數計算得出檢出限;以標準曲線上的最低點,取信噪比為S/N=10,結合稀釋倍數計算得出定量限,試驗結果如表2 所示。8 種氟喹諾酮類藥物的方法檢出限在0.001 μg/kg~0.009μg/kg 范圍內,定量限在0.003 μg/kg~0.031 μg/kg范圍內,均達到了國家標準相關要求。
2.2.3 準確度和精密度
稱取5.00 g 陰性雞蛋樣品,以GB/T 21312—2007 中方法測定低限為基礎,以GB/T 27404—2008《實驗室質量控制規范食品理化檢測》附錄F中檢測方法確認的技術要求為試驗依據,在方法測定低限(1.0 μg/kg)、2 倍方法測定低限(2.0 μg/kg)、10 倍方法測定低限(10.0 μg/kg)進行三水平試驗,按本文優化后的方法進行前處理。8 種氟喹諾酮類藥物的平均回收率為81.60%~103.88%,相對標準偏差為0.78%~9.91%,方法的準確度和精密度均可達到國家標準相關指標要求,具體結果見表2。
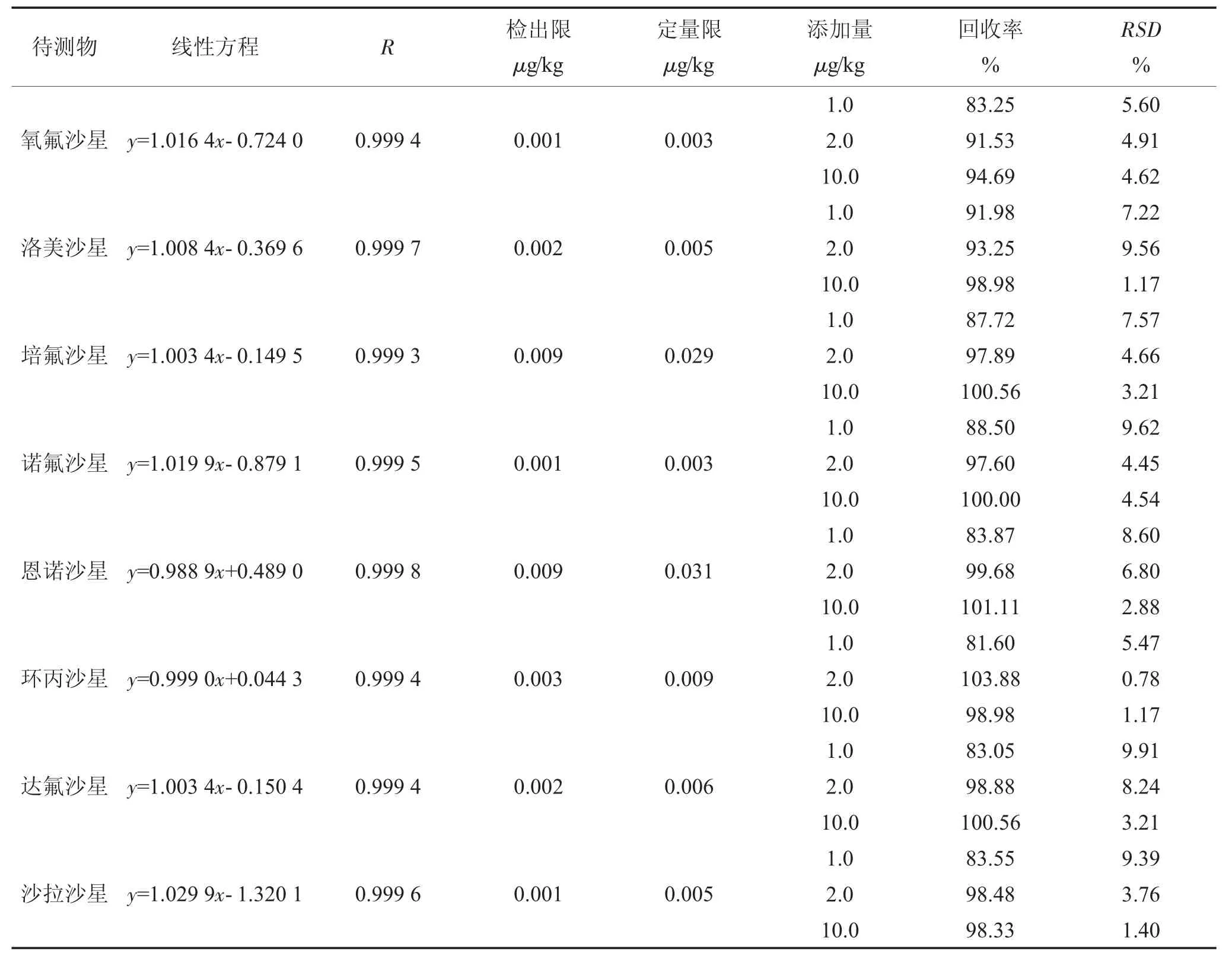
表2 雞蛋中8 種待測物線性方程、相關系數、檢出限、定量限、回收率及精密度(n=6)
3 結論
本試驗對GB/T 21312—2007《動物源性食品中14 種喹諾酮藥物殘留檢測方法液相色譜-質譜/質譜法》中部分步驟進行優化,結果表明,優化后的方法靈敏度更高,重現性更好,能夠滿足雞蛋中氟喹諾酮類藥物殘留的例行檢測。另外,因篇幅有限,文章只提供了針對雞蛋中氟喹諾酮類藥物殘留檢測相關數據,事實上,此方法針對雞肉中的8 種氟喹諾酮類殘留量檢測同樣適用,且回收率和靈敏度均可達到國家標準要求。文章旨在為防范非法添加氟喹諾酮類藥物殘留提供技術依據。
猜你喜歡
中國設備工程(2022年12期)2022-07-11 04:33:00
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:36
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:34
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:50
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:48
