UPLC 法測定大黃蟲丸中5 種蒽醌類活性成分的含量
2021-11-16 08:26:44藍夢柳張海鳳鐘玉環吳水生
福建中醫藥 2021年10期
關鍵詞:蘆薈
卓 輝,藍夢柳,張海鳳,鐘玉環,許 文,吳水生
(福建中醫藥大學藥學院,福建 福州 350122)
1 儀器與材料
1.1 儀器 ACQUITY UPLC H-CLASS 超高效液相色譜儀(美國Water 公司);GT-2120 QTS 智能超聲清洗儀(廣東固特超聲股份有限公司);DV215CD十萬分之一天平(美國OHAUS 公司);ME204E 分析天平(瑞士METTLER 公司)。
2 方法與結果
2.1 混合對照品溶液制備 取蘆薈大黃素、大黃酸、大黃素、大黃酚和大黃素甲醚對照品適量,精密稱定,用甲醇溶解制得質量濃度依次為11.2、14.4、16.0、30.4、12.0 μg/mL 的混合對照品溶液。
2.2 供試品溶液制備 參照2020 年版《中華人民共和國藥典(一部)》[2]大黃蟲丸供試品溶液制備法:取剪碎的大黃蟲丸1 g,精密稱定,置于50 mL具塞錐形瓶中,精密加入甲醇25 mL,稱定重量,浸泡12 h 后用玻棒研磨使樣品溶散,用數滴甲醇沖洗玻棒于錐形瓶中,超聲處理30 min,放冷,再稱定重量,用甲醇補足損失的重量,或揮散至原重量,搖勻,濾過,取續濾液。
2.4 檢測波長的選擇 全波長掃描檢測顯示:蘆薈大黃素、大黃酸、大黃素、大黃酚和大黃素甲醚的最大吸收波長分別為224.7、230.6、222.3、224.7、222.3 nm,但在各個最大吸收波長下,色譜圖均有不同程度的基線漂移。在254 nm 波長下,以上5 種蒽醌類活性成分的色譜圖基線平穩,峰形對稱,故選擇254 nm為檢測波長。
2.5 色譜條件的優化 考察了Syncronis aQ 柱(2.1 mm×100 mm,1.7 μm)、ACQUITY UPLC BEH C18 柱(2.1 mm×100 mm,1.7 μm)和Uitimate UHPLC AQ-C18 柱(2.1 mm×100 mm,1.8 μm)3 種色譜柱對上述5 種成分的分離效果,其中Syncronis aQ 柱(2.1 mm×100 mm,1.7 μm)能較好地分離蘆薈大黃素與大黃酸,且各成分峰形對稱;以甲醇-0.1%甲酸水(85∶15)為流動相時,5 種成分可得到較好分離,但連續進樣后各成分保留時間發生漂移,且靠后的色譜峰峰寬變大,優化為“2.6”項下梯度洗脫條件后,各成分的保留時間未見飄移,且峰寬一致,峰形尖銳。故選擇Syncronis aQ 柱(2.1 mm×100 mm,1.7 μm)色譜柱與甲醇-0.1%甲酸水梯度洗脫的色譜條件。
2.6 色譜條件色譜柱 Syncronis aQ 柱(2.1 mm×100 mm,1.7 μm);流動相:甲醇(A)-0.1%甲酸水(B),梯度洗脫(0~2 min,85%A;2~3 min,85%A→90%A;3~8 min,90%A;8~10 min,90%→85%A);流速:0.20 mL/min;柱溫:30 ℃;進樣量:2 μL;檢測波長:254 nm。
2.7 方法學考察
2.7.1 系統適應性考察 取“2.1”項下混合對照品溶液,按“2.6”項下色譜條件操作,使用Empower 3系統分析蘆薈大黃素、大黃酸、大黃素、大黃酚和大黃素甲醚在254 nm 檢測波長下與其相鄰色譜峰的分離度均大于1.5,理論塔板數均大于8 000。
2.7.2 線性關系考察 取“2.1”項下混合對照品溶液,逐級加甲醇稀釋2、4、8、16、32、64 倍,連同母液共7個濃度,按“2.6”項下色譜條件依次進樣,以峰面積為縱坐標,濃度為橫坐標,繪制標準曲線。將混合對照品逐步稀釋,分別以信噪比等于3 時的樣品濃度為檢測限,以信噪比等于10 時的樣品濃度為定量限。 5 種蒽醌類活性成分的回歸方程、相關系數(r)、線性范圍、檢測限與定量限結果見表1。 表1 表明各成分在其各自質量濃度范圍內呈良好的線性關系。
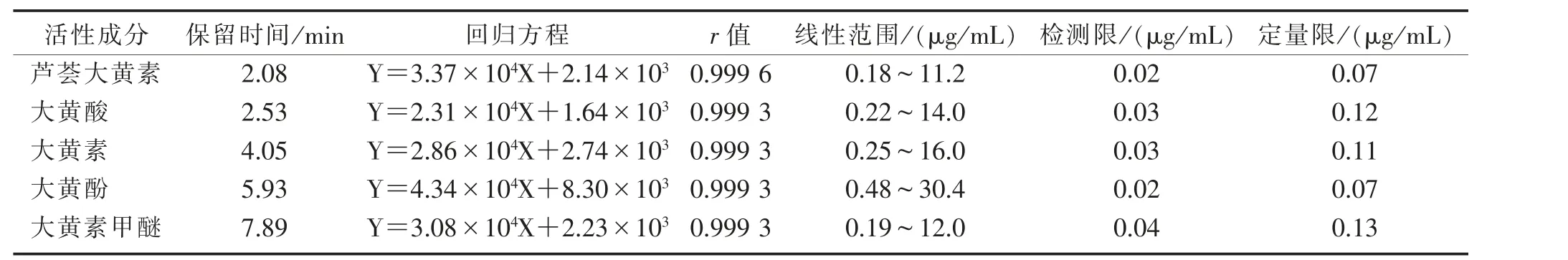
表1 5 種蒽醌類成分的保留時間、回歸方程、線性范圍、檢測限及定量限
2.7.3 專屬性考察 取混合對照品溶液、供試品溶液以及陰性樣品溶液,按“2.6”項下色譜條件進樣檢測,結果見圖1。 混合對照品溶液及供試品溶液中5 種待測成分色譜峰分離度較好,陰性樣品溶液在相應位置未見色譜峰。

圖1 混合對照品溶液、供試品溶液及陰性樣品溶液的UPLC 圖
2.7.4 精密度實驗 取“2.1”項下混合對照品溶液適量,按照“2.6”項下色譜條件連續進樣6 次,記錄色譜圖,以峰面積計算蘆薈大黃素、大黃酸、大黃素、大黃酚和大黃素甲醚色譜峰面積的RSD 分別為1.2%、2.2%、1.4%、0.4%、0.5%,表明儀器的精密度良好。
2.7.5 穩定性實驗 取同一供試品溶液,按照“2.6”項下色譜條件,分別于0、1、2、4、8、12、24 h 進樣2 μL,記錄色譜圖,以峰面積計算蘆薈大黃素、大黃酸、大黃素、大黃酚和大黃素甲醚色譜峰面積的RSD 分別為1.8%、1.9%、2.0%、1.9%、1.7%、2.5%、2.1%,表明供試品溶液在24 h 內穩定。
2.7.7 加樣回收率實驗 取“2.7.6”項下已知含量的大黃蟲丸1.0 g,精密稱定,分別加入相當于樣品中已知5 種蒽醌類化合物質量100%的對照品,按“2.2”項平行制備6 份供試品溶液,按“2.6”項下色譜條件進樣,分別計算5 種蒽醌類成分的加樣回收率,結果見表2。 表2 表明方法準確性較好。

表2 5 種蒽醌類活性成分的加樣回收率實驗結果
表3 不同廠家大黃蟲丸中5 種蒽醌類活性成分的含量測定結果mg/g

表3 不同廠家大黃蟲丸中5 種蒽醌類活性成分的含量測定結果mg/g
廠家北京同仁堂股份有限公司廣東恒誠制藥股份有限公司江蘇頤海藥業有限責任公司生產批號20010357 20010140 20010353 20201007 20210201 20201008 210302 210301 210303蘆薈大黃素0.295 0.263 0.253 0.496 0.450 0.485 0.332 0.357 0.348大黃酸0.805 0.733 0.698 0.184 0.158 0.263 0.496 0.568 0.517大黃素0.448 0.423 0.407 0.205 0.200 0.196 0.389 0.436 0.412大黃酚0.859 0.812 0.784 1.577 1.805 1.539 0.911 0.981 0.959大黃素甲醚0.231 0.219 0.211 0.341 0.386 0.333 0.192 0.206 0.203
表4 不同廠家大黃蟲丸中5 種蒽醌類活性成分的平均總含量與各成分占比
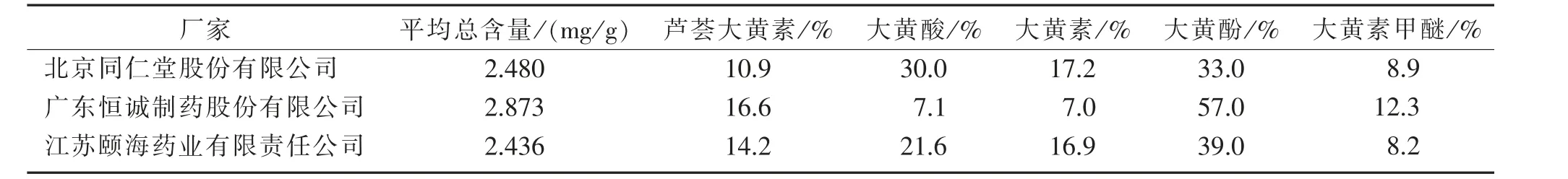
表4 不同廠家大黃蟲丸中5 種蒽醌類活性成分的平均總含量與各成分占比
廠家北京同仁堂股份有限公司廣東恒誠制藥股份有限公司江蘇頤海藥業有限責任公司平均總含量/(mg/g)2.480 2.873 2.436蘆薈大黃素/%10.9 16.6 14.2大黃酸/%30.0 7.1 21.6大黃素/%17.2 7.0 16.9大黃酚/%33.0 57.0 39.0大黃素甲醚/%8.9 12.3 8.2
3 討 論
猜你喜歡
中學生百科·大語文(2023年2期)2023-05-08 14:11:47
故事作文·低年級(2023年1期)2023-02-23 07:19:20
小讀者(2021年6期)2021-07-22 01:50:02
科教新報(2021年23期)2021-07-21 14:37:25
故事作文·低年級(2021年4期)2021-05-06 03:11:13
作文評點報·小學三、四年級(2019年14期)2019-04-30 00:22:22
故事作文·低年級(2016年1期)2016-09-10 07:22:44
故事作文·高年級(2015年5期)2015-09-08 08:27:03
江蘇調味副食品(2015年1期)2015-02-28 01:56:34
故事作文·低年級(2009年5期)2009-05-06 03:35:50
