雙標線性校正法應用于西洋參破壁飲片的指紋圖譜研究△
2021-11-16 07:43:30于現花劉軍玲張亞中金傳山
中國現代中藥 2021年9期
于現花,劉軍玲,張亞中,金傳山
1.安徽中醫藥大學 藥學院,安徽 合肥 230012;2.安徽省食品藥品檢驗研究院,安徽 合肥 230051
西洋參為五加科植物西洋參Panax quinquefoliumL.的干燥根,原產地主要是美國與加拿大,我國從17 世紀開始引進種植。其現已成為我國傳統滋補藥材。西洋參味甘、微苦,性涼,歸心、肺、腎經,具有補氣養陰、清熱生津作用,用于氣虛陰虧、虛熱煩倦、咳喘痰血、內熱消渴、口燥咽干[1]。現代研究發現,西洋參中含有多種有效成分,具有抗腫瘤、抗癌、降血壓、調血脂、抗疲勞等多種藥理活性[2-4]。
西洋參破壁飲片是將傳統飲片細胞壁打破,通過現代超微粉碎技術粉碎加工成D90(樣品累積分布百分數達到90%時所對應的粒徑)<45 μm 的粉體,再用無添加賦形技術制成30~100 目的顆粒新型中藥飲片。破壁飲片可使中藥的有效成分得到更充分釋放,進而大幅提高藥效。與傳統飲片相比,西洋參破壁飲片具有全成分保留,成分利用率高,質量均一,服用方式簡單、快捷、靈活等特點[5-9]。近年來,破壁粉碎技術在中藥領域中的應用日益廣泛,中藥破壁飲片品種逐漸增多,對破壁飲片的評價、產業化及應用等方面的研究已成為中藥傳統飲片傳承與改良創新的熱門方向,并取得了一定成果。
由于外觀性狀及顯微特征已被破壞,傳統飲片的質量評價標準已不適用于破壁飲片質量控制要求[10]。另因西洋參特有成分擬人參皂苷F11為弱紫外吸收成分,因此,需要建立高效液相色譜-電霧式檢測器(HPLC-CAD),用于西洋參傳統飲片與破壁飲片的指紋圖譜分析。目前,指紋圖譜研究較為單一,一般僅在1 根色譜柱上開展分析,后續難以適用到其他色譜柱或用相對保留時間法進行色譜峰的定性,其預測保留時間絕對偏差較大,實用性不佳。本實驗采用雙標線性校正法[11-14]預測特征峰的保留時間,據此建立的西洋參破壁飲片指紋圖譜可以適用于不同色譜柱,既擴展了方法的適用范圍,又降低了對照品的使用成本,為西洋參破壁飲片質量控制提供參考。
1 材料
Ultimate 3000 型高效液相色譜儀(美國Dionex公司);XP26 型百萬分之一電子分析天平(Mettler公司);A11 型分析研磨機(IKA 公司);KQ-100 型超聲儀(昆山市超聲儀器有限公司);Simplicity-185型超純水儀(Millipore公司)。
16根色譜柱,編號col 1~col 16,規格均為250 mm×4.6 mm,5 μm。col 1:Agilent TC-C18(2);col 2:Diamonsil C18;col 3:GL Wondasil C18;col 4:Innoval C18;col 5:Inspire C18;col 6:JADE-PAK ODS-AQ;col 7:Kromasil 100 5-C18;col 8:Luna C18(2)100A;col 9:Shim-pack VP-ODS;col 10:Sonoma C18(2)100A;col 11:Tech Mate C18ST;col 12:Waters Symmetry C18;col 13:ZORBAX Eclipse XDB C18;col 14:ZORBAX SB C18;col 15:Waters XBridge C18;col 16:Kinetex C18100A。
對照品擬人參皂苷F11(批號:110841-200505,純度≥98%)、人參皂苷Rg1(批號:110703-201128,純度≥93.4%)、人參皂苷Re(批號:110754-201626,純度≥92.7%)、人參皂苷Rb1(批號:110704-201122,純度≥92.9%)、人參皂苷Rc(批號:110021-14-0,純度≥98%)、人參皂苷Rb2(批號:111715-200501,純度≥98%)、人參皂苷Rb3(批號:111686-200501,純度≥98%)、人參皂苷Rd(批號:111818-201001,純度≥94.4%)均由中國食品藥品檢定研究院提供;DRS Origin 軟件由中國食品藥品檢驗研究院設計研發,科邁恩(北京)科技有限公司開發;乙腈和甲醇為色譜純;正丁醇為分析純;水為超純水。
10 批西洋參(編號XYS-01~XYS-10)均購于安徽宏信藥業發展有限公司,經安徽省食品藥品檢驗研究院張亞中主任中藥師鑒定為五加科植物西洋參Panax quinquefoliumL.的干燥根。
2 方法與結果
2.1 色譜條件
色譜柱:Waters XBridge C18(250 mm×4.6 mm,5 μm),流動相:乙腈(A)-水(B),線性梯度洗脫(0~5 min,81% B;5~25 min,81%~78%B;25~30 min,78%~74%B;30~55 min,74%~50%B;55~60 min,50%~81%B);柱溫:30 ℃;流速:1 mL·min-1;CAD 霧化器溫度:35 ℃;進樣量:10 μL。上述條件下,各成分分離較好。
2.2 對照品溶液的制備
精密稱取各人參皂苷對照品適量,置于5 mL 量瓶中,加甲醇溶解定容,配制成含人參皂苷Rg1、Re、Rb1、Rc、Rb2、Rb3、Rd 及擬人參皂苷F11分別為0.042 8、0.130 1、0.035 3、0.056 4、0.025 0、0.029 7、0.061 2、0.097 7 mg·mL-1的混合對照品溶液。
2.3 供試品溶液的制備
精密稱取供試品1 g,置具塞錐形瓶中,精密加入甲醇50 mL,超聲(250 W,50 kHz)30 min,放冷,再稱定質量,用甲醇補足減失的質量,搖勻,濾過,取續濾液,即得。
2.4 方法學考察
2.4.1 精密度試驗 取2.2 項下的混合對照品溶液,連續進樣6 次,每次進樣10μL。人參皂苷Rg1、Re、Rb1、Rc、Rb2、Rb3、Rd 及擬人參皂苷F11峰面積的RSD 分別為1.70%、0.30%、0.78%、0.84%、0.49%、0.91%、0.68%、1.55%,表明儀器精密度良好。
2.4.2 重復性試驗 取同一供試品(XYS-02)粉末,分別按2.3 項下方法平行制備6 份供試品溶液,測定并計算其含量。樣品中人參皂苷Rg1、Re、Rb1、Rc、Rb2、Rb3、Rd 及擬人參皂苷F11各含量的RSD分別為0.98%、1.12%、1.08%、1.35%、0.79%、1.02%、1.31%、0.45%,表明本法重復性良好。
2.4.3 穩定性試驗 取新制備的供試品溶液(XYS-02),室溫下放置,分別在0、2、4、6、8、12、24 h 進樣測定,人參皂苷Rg1、Re、Rb1、Rc、Rb2、Rb3、Rd及擬人參皂苷F11峰面積的RSD分別為1.27%、0.96%、1.68%、1.44%、1.64%、0.85%、1.26%、0.43%,表明供試品溶液在24 h內穩定。
2.5 西洋參破壁飲片與傳統飲片相似度評價分析
采用“中藥色譜指紋圖譜相似度評價系統”2012 版軟件,中位數法(時間窗為0.1)分別建立西洋參破壁飲片及傳統飲片指紋圖譜共有模式。結果顯示,西洋參破壁飲片及傳統飲片指紋圖譜均標定17 個共有峰,并指認出1 號峰為人參皂苷Rg1、2 號峰為人參皂苷Re、3 號峰為擬人參皂苷F11、4 號峰為人參皂苷Rb1、5 號峰為人參皂苷Rc、6 號峰為人參皂苷Rb2、7 號峰為人參皂苷Rb3、8號峰為人參皂苷Rd,結果見圖1。計算10 批破壁飲片與傳統飲片之間的相似度。結果顯示,各批破壁飲片與傳統飲片之間的相似度均大于0.939(表1)。可見,不同批次間破壁飲片產品均一性和穩定性較好。由圖2 可知,各批次破壁飲片與傳統飲片之間的關聯性較好,也說明西洋參傳統飲片制成破壁飲片后,各主要成分均較定,體現了破壁飲片與原傳統飲片的物質基礎一致,保留飲片全成分的特性。
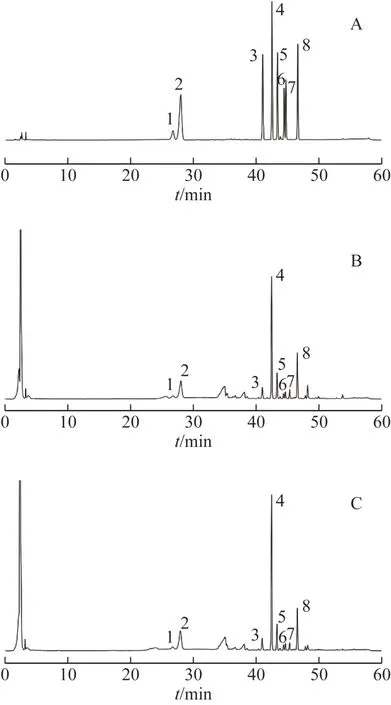
圖1 對照品、西洋參傳統飲片供試品和破壁飲片供試品色譜圖

圖2 西洋參破壁飲片與傳統飲片的指紋圖譜
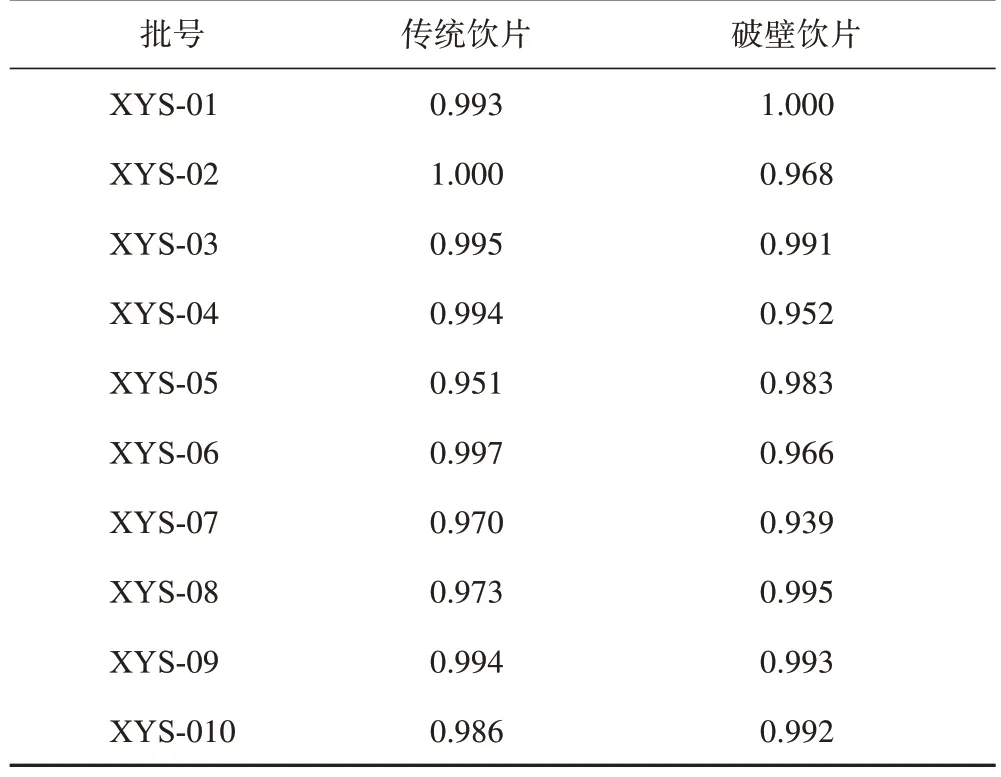
表1 10批西洋參破壁飲片與傳統飲片之間的相似度
2.6 雙標線性校正法定性研究
2.6.1 標準保留時間(SRT)的計算 液相色譜中,化學成分在不同色譜儀和不同色譜柱上的保留時間具有線性關系[15]。通過對照品的保留時間進行色譜峰定位,得到樣品實際保留時間,以15 根色譜柱得到的對照品保留時間平均值作為SRT[16]。分別以8 個成分的SRT 為橫坐標,依次為人參皂苷Rg132.271 min、人參皂苷Re 32.791 min、擬人參皂苷F1142.371 min、人參皂苷Rb143.933 min、人參皂苷Rc 44.881 min、人參皂苷Rb245.833 min、人參皂苷Rb346.142 min、人參皂苷Rd 47.977 min。以實際保留時間為縱坐標,得到各色譜柱的保留時間擬合結果,見圖3,各色譜柱的線性回歸方程和相關系數見表2。

圖3 不同色譜柱保留時間關系
由圖3 和表2 可見,col 15 色譜柱的保留時間擬合直線相對有偏離,說明成分在col 15色譜柱相較于其他色譜柱保留時間相差較大。余下col 1~col 14 色譜柱相關系數均在0.999 以上,說明這14 根色譜柱的保留時間的線性關系良好,可用于西洋參破壁飲片的定性評價分析。

表2 不同色譜柱上保留時間的線性方程及相關系數
2.6.2 雙標化合物的確定 通過DRS Origin 軟件的計算,篩選出了10種雙標選擇方案(表3),10種方案色譜柱符合率均達到95%,其中峰2~6 組合的預測正確率及色譜柱符合率均為100%,且保留時間回歸偏差最小。在未知色譜柱(以col 16 為驗證柱)上運行雙標對照品溶液和供試品溶液,將人參皂苷Re 和人參皂苷Rb2作為西洋參破壁飲片液相色譜的雙標化合物,以這2 個成分的SRT 為橫坐標,實際保留時間人參皂苷Re 32.791 min、人參皂苷Rb245.833 min 為縱坐標,得到2 點人參皂苷Re(32.791、31.628)和人參皂苷Rb2(45.833、44.491),做一直線,得到方程Y=0.986 3X-0.712 9,然后將其余6 個成分的SRT 值代入方程,得到SRT:人參皂苷Rg131.116 min、擬人參皂苷F1141.078 min、人參皂苷Rb142.618 min、人參皂苷Rc 43.553 min、人參皂苷Rb344.797 min、人參皂苷Rd 46.607 min。實際保留時間依次為31.628、41.036、42.828、43.663、44.757、46.451 min。單純定性條件下,滿足預測保留時間與實測保留時間的誤差應不超過0.5 min 的要求[16],說明該雙標選擇在col 16 上保留時間預測效果良好。

表3 10種雙標化合物選擇方案
2.6.3 雙標線性校正法與相對保留時間法的對比對雙標線性校正法與相對保留時間法在西洋參破壁飲片液相色譜中定性的優劣進行比較。雙標線性校正法以色譜圖靠近兩端的人參皂苷Re和人參皂苷Rb2為雙標化合物,相對保留時間法以人參皂苷Rb2為參照物,表4 為在15 根色譜柱上2 種方法的比較結果,與相對保留時間法比較,雙標線性校正法的保留時間預測值的絕對偏差波動范圍較小且絕對偏差較低。由此可見,雙標線性校正預測保留時間的準確性優于相對保留時間。
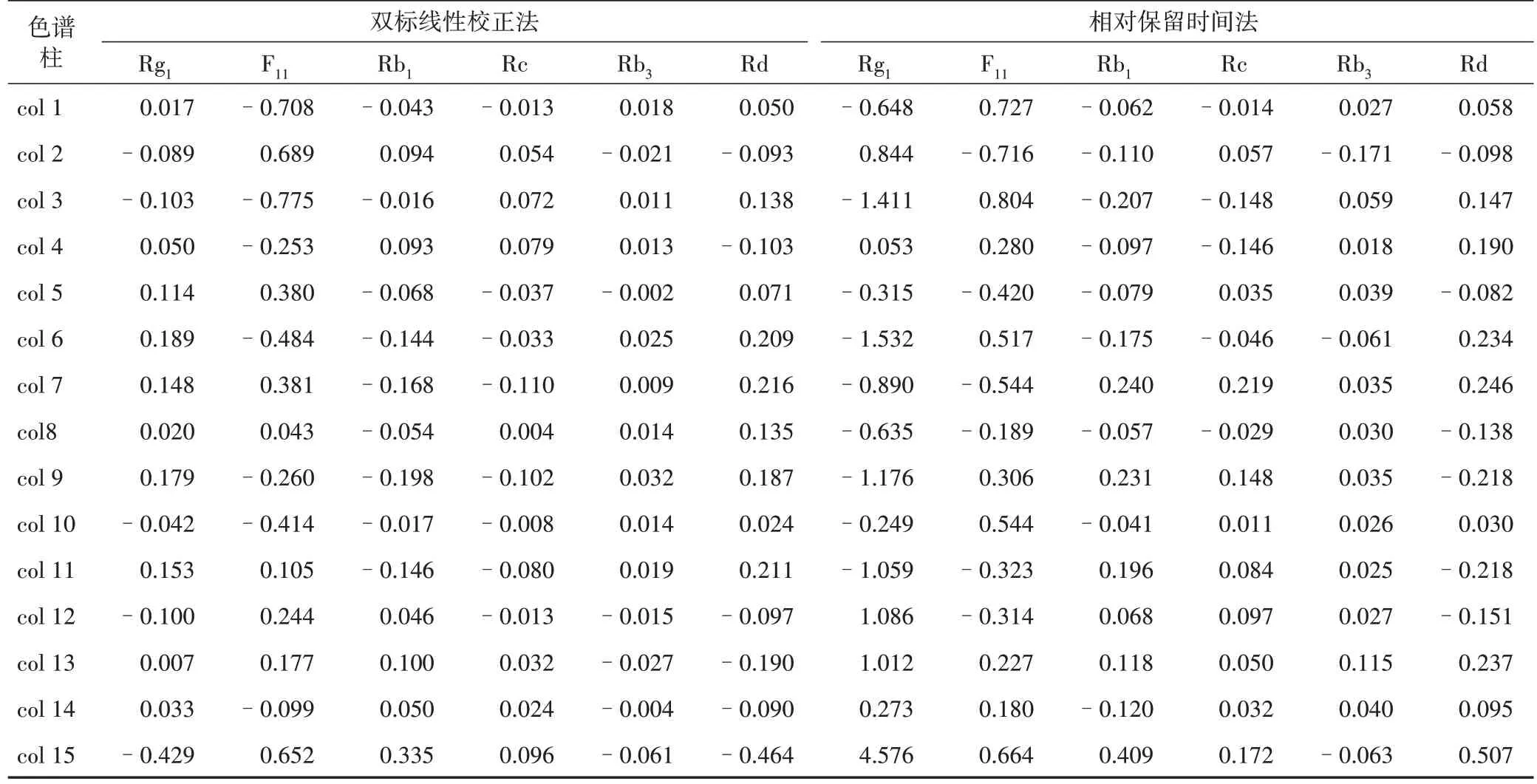
表4 不同色譜柱2種方法不同成分保留時間預測值的絕對偏差
3 討論
3.1 檢測器的選擇
擬人參皂苷F11作為西洋參特征成分,母體結構為奧克娣隆型化合物,在紫外區僅有末端吸收。通過比較西洋參破壁飲片的CAD 與二極管陣列檢測器(DAD)檢測圖譜,發現DAD 下未見相應峰,而CAD 下響應較好,指紋特征性更強且靈敏度高,基線更平穩。故選擇CAD 建立西洋參破壁飲片與傳統飲片的指紋圖譜研究。
3.2 前處理及色譜條件的選擇
本實驗選用HPLC 測定皂苷類成分,對水飽和正丁醇與甲醇不同溶劑、超聲與加熱回流不同提取方法及提取時間等進行考察發現,甲醇超聲30 min提取的供試品溶液色譜峰信息全面完整且操作簡單,足以適用于對西洋參破壁飲片定性研究。所用流動相有乙腈-0.01%三氟乙酸、乙腈-0.1%甲酸、乙腈-水等[17-18]。經實驗比較發現,以乙腈-水進行梯度洗脫系統顯示的圖譜峰形好、基線平穩,可滿足8 個皂苷類成分的分離要求,且水相較于三氟乙酸和甲酸更安全、經濟又方便。
本研究采用HPLC-CAD 建立西洋參破壁飲片的指紋圖譜,同時,對同批次來源的西洋參傳統飲片指紋圖譜的相似度進行評價分析,探究兩者之間的關聯性。結果顯示,傳統飲片與破壁飲片之間色譜峰相似度均大于0.939,各共有峰的保留時間幾乎一致。這表明,西洋參傳統飲片經破壁粉碎并制成破壁飲片的過程中,化學成分無明顯變化,超微破壁粉碎技術并未造成傳統飲片中主成分流失及新化學成分的轉化或生成[19]。筆者又結合DRS Origin 軟件對15 根色譜柱采集的西洋參破壁飲片數據擬合,進而對色譜峰定位,確定雙標化合物,再用雙標線性校正預測目標化合物的保留時間。結果與相對保留時間法相比,雙標線性校正法預測定位更準確且實用方便,表明該方法在中藥復雜的多指標成分定性方面有著很好的應用前景。
