高溫大曲中吡嗪類化合物GC-MS檢測方法的研究
2021-11-24 07:24:10呂志遠秦炳偉張夢夢邱振清韓云翠時雪萌王瀅瀅呂洪霞
釀酒科技 2021年11期
關鍵詞:檢測
蘇 寧,呂志遠,秦炳偉,張夢夢,邱振清,韓云翠,時雪萌,王瀅瀅,呂洪霞
(濟南趵突泉釀酒有限責任公司,山東濟南250115)
吡嗪是1,4位含氮的六元環化合物,具有明顯的焦香或炒堅果、炒芝麻的香氣[1]。白酒中吡嗪類化合物具有閾值低,氣味強度高的特點,對焦香的形成具有很重要的作用,是白酒中重要的風味物質,也是大曲揮發性香味物質的重要組成成分。大曲在制作過程中產生的各類香味物質,可隨著發酵和蒸餾過程進入酒體,因此,大曲中的香味物質對白酒的風味特征和品質具有一定的貢獻[2]。目前,對于吡嗪類化合物的研究主要集中于對白酒中該類化合物的檢測與分析[3],對大曲中吡嗪類化合物的檢測方法研究較少,牛姣等[4]通過頂空固相微萃取結合氣相色譜質譜聯用技術,對仰韶陶香型高溫大曲中揮發性香味物質進行分析,共檢測出4種吡嗪類化合物。本研究采用GC-MS對高溫大曲中吡嗪類化合物進行檢測,共檢出7種含一個或者多個甲基的吡嗪類化合物,為今后高溫大曲的香味物質及最佳儲存期的研究奠定了基礎。
1 材料與方法
1.1 材料、試劑及儀器
材料:選用培曲期間(第0天~25天)及前期儲存(第26天~80天)的高溫大曲。
試劑及耗材:無水乙醇(色譜純);內標物,2-甲氧基-3-甲基吡嗪;吡嗪類標準品,2-甲基吡嗪、2-乙基吡嗪、2,3-二甲基吡嗪、2,3-二乙基吡嗪、2,3,5-三甲基吡嗪、四甲基吡嗪、2,5/2,6-二甲基吡嗪。內標物與標準品,純度均為99.9%,購自百靈威科技有限公司。
儀器設備:氣相色譜質譜聯用儀(7890 A-5975 C),安捷倫科技有限公司;電子天平(精確到0.1 g);分析天平(型號FA 2004,精確到0.0001 g),天津天馬衡基儀器有限公司;超聲波發生器(KQ 2200型),昆山市超聲儀器有限公司。
1.2 試驗方法
1.2.1 試驗準備
內標標準儲備液:稱取0.01 g(精確至0.0001 g)的2-甲氧基-3-甲基吡嗪,使用60%乙醇水溶液溶解定容至100 mL,濃度為100 mg/L。
內標使用液:準確吸取10 mL內標標準儲備液,使用60%乙醇水液稀釋并定容至100 mL,濃度為10 mg/L。
吡嗪類混合標準儲備液:分別稱取0.1 g(精確至0.0001 g)吡嗪類標準品,使用60%乙醇水溶液溶解后定容至100 mL,使標準儲備液濃度為1000 mg/L。
吡嗪類混合標準工作液:分別準確吸取2,5/2,6-二甲基吡嗪標準儲備200 μL,其余標準儲備液100 μL,使用60%乙醇水溶液定容至100 mL,使2,5/2,6-二甲基吡嗪的濃度為2 mg/L,其余吡嗪類濃度為1 mg/L。配制標準曲線濃度:2,5/2,6-二甲基吡嗪濃度梯度為0.2 mg/L、0.4 mg/L、1 mg/L、1.6 mg/L、2 mg/L,其余吡嗪類濃度梯度為0.1 mg/L、0.2 mg/L、0.5 mg/L、0.8 mg/L、1.0 mg/L,且內標濃度均為1 mg/L,待測。
1.2.2 試驗前處理及優化
樣品前處理:稱取10 g(精確至0.1 g)大曲于三角瓶中,加入45 mL乙醇水溶液和5 mL內標使用液,超聲,離心取上層清液,0.45 μm濾膜過濾,準確吸取1 mL濾液至進樣瓶中,供GC-MS分析。
分別采用15%、25%、50%、60%乙醇水溶液作為提取溶劑,確定最優提取溶劑濃度。
加入乙醇水溶液后,分別采用20 min、30 min、60 min超聲提取吡嗪類化合物,確定最優超聲提取時間。
1.2.3 GC條件
色譜柱為HP-5 MS(30 m×0.25 mm×0.25 μm);進樣口溫度為250℃;載氣為氦氣,流速1 mL/min;程序升溫起始溫度為40℃,保留5 min,然后以3.5℃/min升至90℃,再以15℃/min升至270℃,保留5 min;分流進樣,分流比10∶1,分流流量10 mL/min,進樣量1 μL。
1.2.4 MS條件
電子轟擊離子源(EI);電子能量70 eV;傳輸線溫度280℃;離子源溫度230℃。
1.2.5 定性定量方法
定性:SCAN模式下采集圖譜,利用NIST 08標準譜庫定性,確定監測離子參數。
定量:根據SCAN檢測模式得到的離子檢測參數,建立SIM檢測模式,采用內標與標準曲線法,對吡嗪類化合物定量,每個樣品測量3次。
1.2.6 方法檢出限、回收率及精密度實驗
以信噪比S/N為3計算出方法檢出限。利用優化的前處理方法,進行回收率與精密度實驗:分別取10 g大曲(同一個樣品)于三角瓶中,加入45 mL乙醇水溶液和5 mL內標使用液,然后添加不同濃度的吡嗪類化合物標品,其中,2,5/2,6-二甲基吡嗪添加水平分別為0.4 mg/L、1 mg/L、2 mg/L,其余吡嗪類添加水平分別為0.2 mg/L、0.5 mg/L、1 mg/L,超聲,離心取上層清液,0.45 μm濾膜過濾,準確吸取1 mL濾液至進樣瓶中,待測,作為加標回收實驗;每個添加水平分別做6個平行樣品,計算相對標準偏差,作為該方法的精密度。
2 結果與分析
2.1 標品和樣品的定性檢測
定性分析:對大曲樣品圖譜的每個峰,利用NIST 08檢索每個吡嗪類化合物,然后利用2 mg/L 2,5/2,6-二甲基吡嗪和1 mg/L其余吡嗪類的標準品,對保留時間和定性離子驗證,最后確定了大曲中7種吡嗪類化合物,其中保留時間和定性離子見表1。
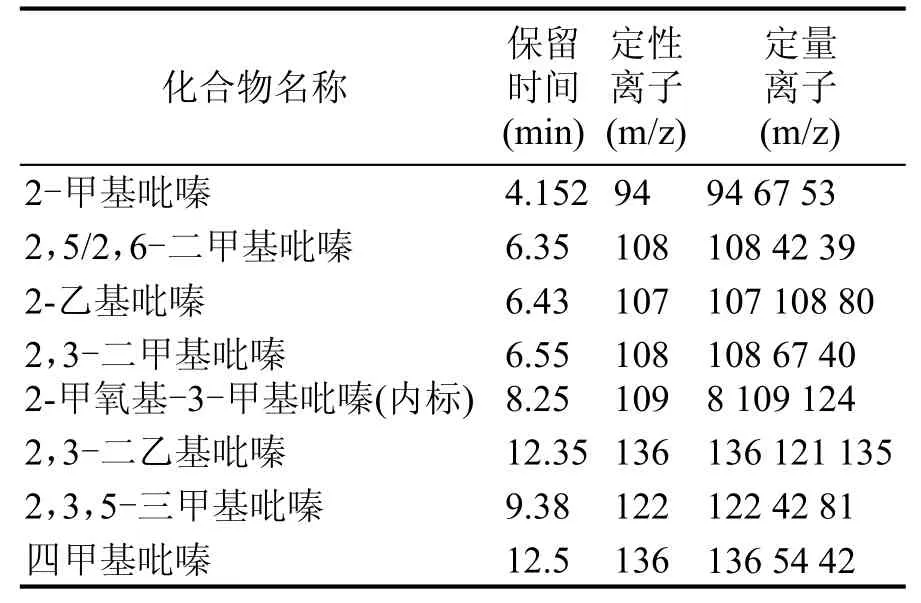
表1 7種吡嗪類化合物監測離子參數
定量分析:通過表1的定性分析,確定了7種吡嗪類化合物的定量離子,通過保留時間與定量離子,建立7種吡嗪類化合物的SIM檢測模式,根據內標法,建立吡嗪類化合物的標準曲線,其中,大曲樣品色譜圖見圖1。
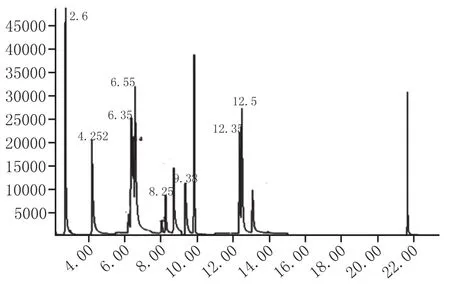
圖1 大曲樣品7種吡嗪類化合物和內標的色譜圖
2.2 前處理方法的優化
2.2.1 提取液的優化
分別采用蒸餾和浸泡的方式對大曲吡嗪類物質進行提取。與浸泡方式相比,蒸餾過程極易產生大量氣泡,導致液體溢出,危險且造成提取物的損失。
浸泡溶劑濃度優化:色譜柱為弱極性色譜柱,用水作為提取劑的樣液,經過色譜柱時,會有一定的柱流失,影響色譜柱的壽命。結合柱損與乙醇(色譜級)價格,試驗初步定乙醇水溶液作為提取溶劑,其濃度分別為15%、25%、50%和60%,對同一大曲樣品浸泡超聲30 min提取,檢測過程相同,結果對比見圖2。
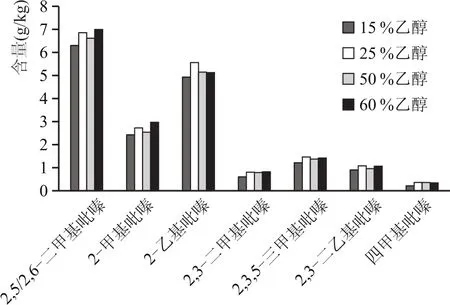
圖2 同一曲樣不同濃度乙醇水溶液吡嗪類化合物檢測結果
由圖2可知,同一大曲樣品在25%、50%、60%乙醇水溶液分別作為提取溶劑時,7種吡嗪類化合物的檢測結果相差較小,而15%乙醇水溶液作為提取溶劑,檢測結果含量最低。因此,選用濃度較低的25%乙醇水溶液對大曲進行浸泡處理最優。
2.2.2 超聲時間優化
用25%乙醇水溶液,提取同一大曲樣品中吡嗪類化合物,超聲時間分別為20 min、30 min、60 min,檢測過程相同,結果對比見圖3。
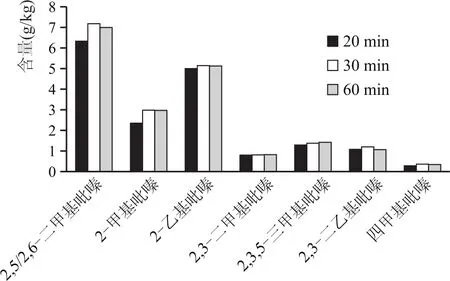
圖3 同一曲樣不同超聲時間吡嗪類化合物檢測結果
由圖3可看出,對同一樣品、同一乙醇水溶液浸泡,不同超聲時間提取對比結果發現,30 min與60 min超聲對比,檢測結果相差不大;與20 min超聲相對比,檢測結果含量明顯高于20 min超聲結果含量。因此,30 min是最優超聲時間。
對標準溶液進行檢測分析,系列標準溶液中各組分含量(mg/L)與內標含量(mg/L)的比值為X橫坐標,吡嗪類各組分與其內標的峰面積的比值為Y縱坐標,得到7種吡嗪類化合物的標準曲線,其線性方程、線性范圍和相關系數見表2。
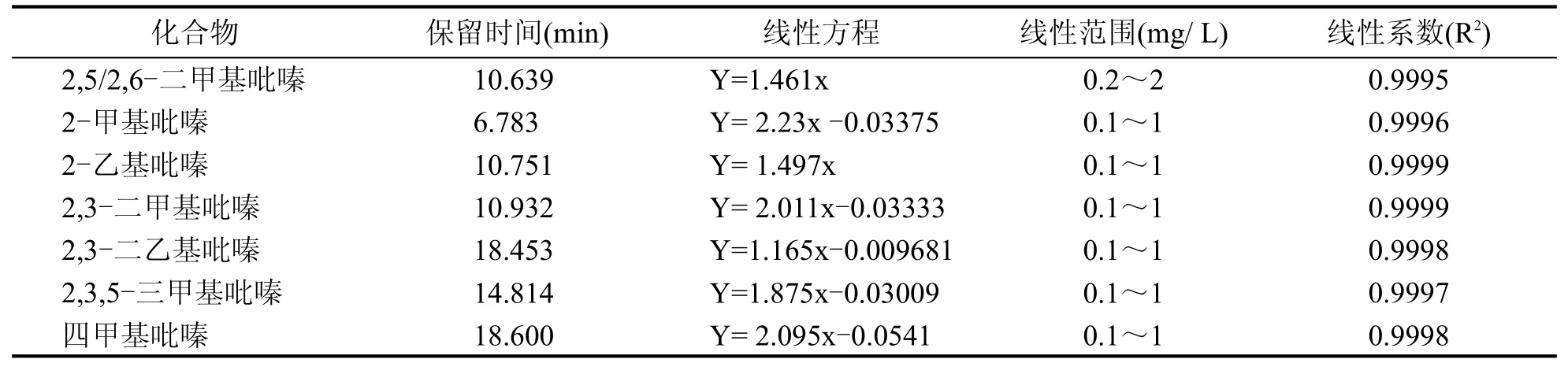
表2 7種吡嗪類化合物線性方程、線性范圍和相關系數
方法檢出限、試驗回收率與精密度,結果見表3。
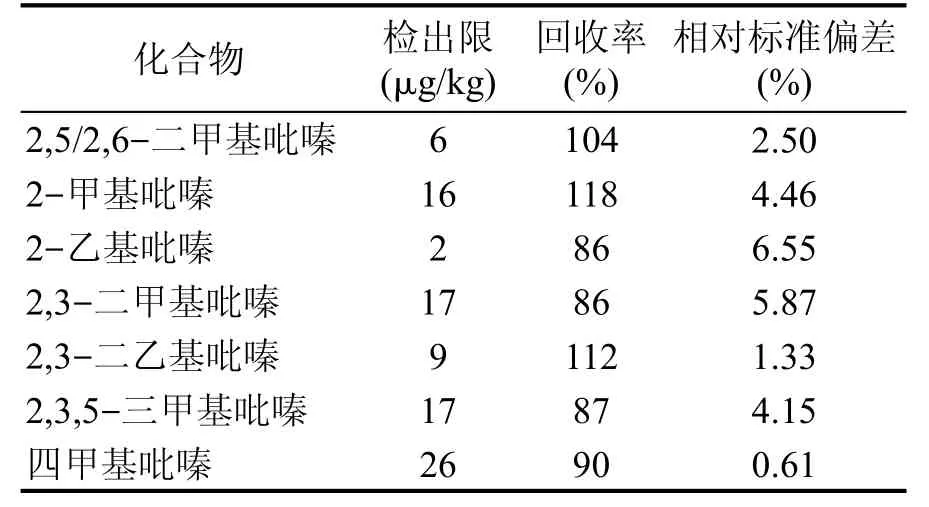
表3 方法檢出限、回收率、相對標準偏差
由表2、表3可得,7種吡嗪類化合物線性相關系數均大于0.999,方法檢出限在2~26 μg/L之間,加標回收率為86%~118%,相對標準偏差為0.61%~6.55%。說明該方法適用于高溫大曲中7種吡嗪類化合物的檢測。
2.4 樣品檢測驗證分析
按照試驗方法對培曲及前期儲存期間的高溫大曲樣品進行檢測,其7種吡嗪類化合物含量的動態變化見圖4。

圖4 7種吡嗪類化合物在培曲及前期儲存期間的變化曲線
由圖4可得,7種吡嗪類化合物在培曲及前期儲存期間的高溫大曲中均被檢測出。其中,2,5/2,6-二甲基吡嗪的含量均明顯高于其他6種吡嗪類化合物,四甲基吡嗪的含量最低;隨發酵天數的增長,這7種吡嗪類化合物的總含量也不斷增加。吡嗪類化合物主要由芽孢桿菌屬代謝產生[5-7],梁晨等[8]對大曲儲存過程中原核生物群落結構及風味成分演替規律研究表明,儲存前期芽孢桿菌屬在高溫大曲中的含量增加,這也與本實驗的結果符合。
3 結論
通過優化試驗方法,采用GC-MS技術,建立了一種高溫大曲中7種吡嗪類化合物的檢測方法,本方法用25%乙醇水溶液,超聲30 min對大曲進行浸泡處理,既最大程度的減少了目標化合物的損失,也節約了成本,同時,該方法操作簡單,檢出限低,靈敏度可靠,適用于高溫大曲中吡嗪類化合物的檢測。
培曲期間(0~25 d)及前期儲存(第26天~80天)的高溫大曲,7種吡嗪類化合物的總含量在第80天時達到最高,可達5.455 mg/kg,其中,2,5/2,6-二甲基吡嗪的含量最高,均明顯高于其他6種吡嗪類化合物。本實驗只對0~80 d的高溫大曲進行了檢測,所以高溫大曲中7種吡嗪類化合物的具體變化規律及最佳儲存期的選擇需進一步擴大范圍進行研究。
猜你喜歡
中國設備工程(2022年12期)2022-07-11 04:33:00
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:36
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:34
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:50
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:48
