季銨鹽聚砜/聚乙烯醇/單寧酸復合水凝膠的制備及性能
2021-11-26 07:49:16楊明成張本尚張秋霞劉樹博郭文慧
吉林大學學報(理學版) 2021年6期
關鍵詞:質量
楊明成, 陳 陽, 張本尚, 張秋霞, 劉樹博, 郭文慧
(1. 河南省科學院 同位素研究所有限責任公司, 河南省輻射新材料工程技術研究中心, 鄭州 450015; 2. 河南省眼健康產品工程技術研究中心, 鄭州 450003)
水凝膠是一種可吸收和保持大量水分的三維交聯聚合物網絡, 自Wichterle等[1]首次報道以來, 水凝膠材料已引起人們廣泛關注, 在組織工程支架、藥物輸送系統、軟性接觸鏡、防污涂層 、傳感器、執行器、軟機器人、廢水處理和傷口敷料等領域應用廣泛[2-13].
聚乙烯醇(PVA)作為一種親水性可生物降解的聚合物, 由于其優異的生物相容性和對溫度變化的穩定性以及對活體組織和細胞的無毒性, 因此廣泛用于水凝膠的制備[14-17]. 聚砜(PSF)是一種性能優異的商品化聚合物, 具有熱穩定性高、耐水解、無毒、耐輻射及生物相容性良好等特點. 但PSF在水中基本不溶解, 需要溶解在水溶性的有機溶劑中, 與PVA水溶液混合后在其他輔助試劑存在的條件下制備PSF/PVA水凝膠, 但有生物毒性的有機溶劑和輔助試劑易殘留在水凝膠中, 難以除去. Muya等[18]將PSF溶于N,N-二甲基甲酰胺(DMF)中, 攪拌條件下加入PVA粉末和戊二醛交聯劑溶液, 在鹽酸作為催化劑75 ℃條件下反應3 h, 并在室溫下保存10 d完成交聯反應, 制備了PSF/PVA水凝膠, 但制備過程中使用了DMF和交聯劑, 后續難以處理. Mbambisa等[19]在鹽酸和N-羥基丁二酰亞胺(NHS)存在的條件下合成了PSF/PVA水凝膠, 該方法制備水凝膠也殘留有機溶劑和NHS. 與常規方法相比, 輻射法制備水凝膠無需使用具有生物毒性的引發劑、交聯劑或其他輔助試劑, 可簡化制備工藝和降低成本, 并可得到相對純凈的水凝膠[20-23].
為解決PSF/PVA復合水凝膠制備過程中使用有機溶劑(如DMF)和其他助劑(交聯劑和催化劑等)的殘留問題, 本文研究了季銨鹽聚砜和聚乙烯醇(QAPSF/PVA)水凝膠的制備方法及性能, 以PSF為原料制備高取代度的氯甲基化聚砜(CMPSF), 并與三甲胺水溶液反應, 經透析、冷凍干燥得到水溶性的QAPSF固體; 將QAPSF和PVA的水溶液混合均勻后, 經60Co γ射線, 制備QAPSF/PVA水凝膠; 將水凝膠浸泡單寧酸(TA)后, 得到了季銨鹽聚砜/聚乙烯醇/單寧酸(QAPSF/PVA/TA)水凝膠; 為研究QAPSF/PVA/TA水凝膠中的氫鍵相互作用, 采用美國Accelrys公司的Materials Studio (MS) 7.0模擬軟件, 以COMPASS(condensed-phase optimized molecular potentials for atomistic simulation studies)為力場, 各組分的最小重復單元為基礎建模, 對QAPSF/PVA/TA水凝膠中分子間的氫鍵相互作用進行模擬計算.
1 實 驗
1.1 試劑與儀器
聚乙烯醇(PVA, 聚合度1 750±50), 天津大茂化學試劑廠; 單寧酸(TA), 分析純, 天津大茂化學試劑廠; 聚砜(PS-3500LCD, MW=77 000), 蘇威特種聚合物公司; 三甲胺(體積分數為30%水溶液), 國藥集團化學試劑有限公司; 無水乙醇, 分析純, 天津大茂化學試劑廠; 大腸桿菌ATCC 25922、金黃色葡萄球菌ATCC 25923, 美國典型培養物保藏中心.
核磁共振波譜儀Agilent-NMR-vnmrs 400型(美國安捷倫公司);60Co 輻射源活度7.4×1015Bq(中核同興(北京)核技術有限責任公司); 萬能試驗機AGS-X-5KN型(日本島津公司); Fourier變換紅外光譜儀Nicolet iS10型(美國賽默飛世爾科技公司); 熱重-差熱聯用熱分析儀SDT-Q600型(美國TA公司); 紫外-可見分光光度計(北京萊伯泰科儀器有限公司); 電子掃描顯微鏡JSM-7001F型(日本電子株式會社).
1.2 水溶性季銨鹽聚砜及水凝膠樣品的制備
參考文獻[24] 制備高取代度的氯甲基化聚砜. 在氮氣氛圍下將100 g聚砜溶于5 000 mL無水氯仿中, 攪拌下依次加入多聚甲醛90 g、三甲基氯硅烷500 mL和無水四氯化錫8 mL, 加熱至50 ℃, 用核磁共振氫譜(1H NMR)檢測跟蹤反應, 當氯甲基化取代度大于1.5時, 停止反應, 將反應液倒入大量乙醇中沉淀析出, 過濾、干燥. 取一定量高取代度的氯甲基化聚砜和100 mL體積分數為15%的三甲胺水溶液放入反應瓶中, 40 ℃下攪拌24 h得到季銨鹽聚砜, 減壓除去大部分三甲胺, 經截留分子量為1 000的透析袋透析除去殘留的三甲胺, 最后冷凍干燥得到白色的季銨鹽聚砜固體.
稱取10 g PVA于70 mL純水中, 90 °C下攪拌至完全溶解得到透明的PVA水溶液, 降溫至60 ℃, 加入質量分數為10%的QAPSF水溶液20 g, 攪拌均勻倒入30 mm×20 mm×2 mm的玻璃模具中, 置于真空干燥箱中抽真空, 并設置溫度為70 ℃, 靜置消除氣泡; 在輻射劑量率為3.09 kGy/h處, 室溫下經60Co輻射源輻射后即可得到透明的QAPSF/PVA水凝膠.
將QAPSF/PVA水凝膠浸泡在純水和不同質量分數(0.1%,0.2%,0.3%,0.4%)的單寧酸溶液中至溶脹平衡, 得到QAPSF/PVA/TA水凝膠.
1.3 測試及表征
1.3.1 取代度的測定
氯甲基化聚砜的取代度可通過1H NMR中相應的積分面積計算, 取代度的計算公式為

(1)
其中DS為取代度,SB為氯甲基中質子峰的積分面積,SA為聚砜骨架中甲基質子峰的積分面積.
1.3.2溶脹性能
將一定質量的QAPSF/PVA水凝膠分別在純水和不同質量分數的(0.1%,0.2%,0.3%,0.4%)TA溶液中浸泡24 h, 每隔一段時間將水凝膠取出, 用濕濾紙擦干其表面水分進行稱質量, 記為Wt. 水凝膠溶脹度的計算公式為
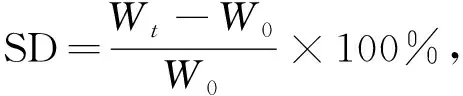
(2)
其中SD為溶脹度(%),W0為水凝膠浸泡前的質量(g),Wt為水凝膠在純水和TA溶液中浸泡后的質量(g).
1.3.3 透明性測試
將QAPSF/PVA/TA水凝膠剪為4 mm×15 mm的長條狀樣品, 置于比色皿中, 用紫外-可見分光光度計測試其透明性.
1.3.4 力學性能測試
拉伸性能測試. 將QAPSF/PVA/TA水凝膠剪為啞鈴形樣條, 用萬能實驗機測試其拉伸性能, 拉伸速率50 mm/min, 樣條有效尺寸為30 mm×4 mm×2 mm, 5個樣品一組.
壓縮性能測試. 將QAPSF/PVA/TA水凝膠剪為圓柱形, 用萬能試驗機測試其壓縮性能, 壓縮速率1 mm/min, 樣品直徑為16 mm, 厚度2 mm, 5個樣品一組.
1.3.5 掃描電子顯微鏡(SEM)表征
將冷凍干燥后的QAPSF/PVA/TA水凝膠樣品在液氮中脆斷, 對斷面噴金處理后, 用掃描電子顯微鏡(SEM)表征斷面形貌.
1.3.6 抑菌性測試
按GB 15979-52002中附錄C有關非溶出性抗(抑)菌產品抑菌性能試驗方法進行測試.
將0.75 g樣品置于250 mL的三角燒瓶中, 分別加入70 mL PBS和5 mL菌懸液, 使菌懸液在PBS中的濃度為1×104~9×104cfu/mL(cfu為菌落數). 將三角燒瓶固定于振蕩搖床上, 以300 r/min振搖1 h. 取0.5 mL振搖后的樣液和用PBS作適當稀釋后的樣液, 以瓊脂傾注法接種平皿, 進行菌落計數. 同時設對照樣片組和不加樣片組, 對照樣片組的對照樣片與被試樣片大小相同但不含抗菌成分, 其他操作程序與被試樣片組均相同, 不加樣片組分別取5 mL菌懸液和70 mL PBS加入同一250 mL三角燒瓶中, 混勻, 分別于0時刻和振蕩1 h后, 各取0.5 mL菌懸液與PBS的混合液做適當稀釋, 然后進行菌落計數. 重復實驗3次, 按

(3)
計算抑菌率, 其中X為抑菌率(%),A為被試樣品振蕩前的平均菌落數,B為被試樣品振蕩后的平均菌落數. 被測試樣品與對照樣品抑菌率的差值X>26%, 可判定被測試樣品具有抑(抗)菌性.
2 結果與討論
2.1 反應時間對CMPSF取代度的影響
提高季銨鹽抑菌官能團在高分子鏈上的密度可提高聚砜高分子材料的抑菌性, 在物料比一定的條件下, 可通過延長反應時間得到取代度高的CMPSF. 表1為反應時間對CMPSF取代度的影響. 由表1 可見, 隨著反應時間的增加, 取代度DS逐漸增加, 當反應時間為72 h時, DS達到最大1.6; 繼續增加反應時間, DS基本無變化, 無法達到DS=2的最大理論取代度, 反而增加了氯甲基聚砜分子鏈之間或分子鏈內部發生聚合反應的幾率, 反應液略微渾濁, 導致后續制備季銨鹽聚砜的水溶性變差. 因此, 在上述物料比一定的條件下, 最佳反應時間為72 h. 反應結束后與三甲胺水溶液反應, 經透析、冷凍干燥得白色季銨鹽聚砜固體. 圖1為QAPSF的1H NMR譜. 由圖1可見, 苯環上的質子峰在化學位移δ=7.87~6.83出現; 與N相連的亞甲基即氯甲基中的質子峰在化學位移δ=4.5附近裂分為兩個峰; 當氯甲基取代度大于1時, 季銨化后可能因為空間位阻或其他原子的影響, 使得與N相連的亞甲基中的質子裂分為左右不對稱的雙峰; 與N相連的—CH3中質子峰在化學位移δ=3.14~3.04出現; 聚砜骨架中雙酚A中—CH3的質子峰化學位移δ=1.70.

表1 反應時間對CMPSF取代度的影響

圖1 QAPSF的1H NMR 譜Fig.1 1H NMR spectrum of QAPSF
2.2 吸收劑量對QAPSF/PVA水凝膠的影響
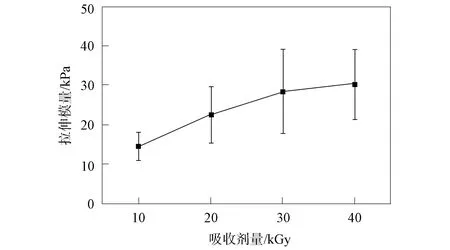
圖2 吸收劑量對QAPSF/PVA水凝膠拉伸模量的影響Fig.2 Effect of absorbed dose on tensile modulus of QAPSF/PVA hydrogels
圖2 為吸收劑量對QAPSF/PVA水凝膠拉伸模量的影響. 由圖2可見, 吸收劑量為30 kGy和40 kGy得到QAPSF/PVA水凝膠的拉伸強度基本一致, 因此選擇吸收劑量為30 kGy. TA與聚乙烯醇可形成較強的氫鍵[25-29], 因此, 將QAPSF/PVA水凝膠浸泡在TA溶液中, 有助于在水凝膠中形成氫鍵以提高其拉伸強度, 并可作為緩釋的抑菌劑提高水凝膠的抑菌性能.
2.3 TA對QAPSF/PVA水凝膠的拉伸和壓縮性能影響
圖3為TA溶液的質量分數對QAPSF/PVA水凝膠拉伸性能的影響. 由圖3可見, QAPSF/PVA水凝膠的拉伸強度和斷裂伸長率均隨TA質量分數的增加呈先增大后減小的趨勢, 且在w(TA)=0.2%時達到最大, 分別為129.1 kPa和177.6%, 與浸泡在純水中的水凝膠(28.3 kPa和80.9%)相比, 在w(TA)=0.2%溶液浸泡后的水凝膠拉伸強度和斷裂伸長率分別提高了4.6倍和2.2倍.
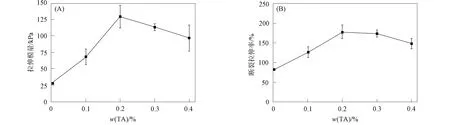
圖3 不同質量分數的TA溶液對QAPSF/PVA水凝膠拉伸性能的影響Fig.3 Effect of TA solution with different mass fraction on tensile properties of QAPSF/PVA hydrogels
圖4為不同質量分數TA溶液對QAPSF/PVA水凝膠壓縮性能的影響. 由圖4可見, QAPSF/PVA水凝膠的壓縮模量隨TA質量分數的增加呈先增大后減小的趨勢. 當w(TA)=0.2%時, QAPSF/PVA水凝膠的壓縮模量達0.49 MPa, 是純水中浸泡水凝膠(0.39 MPa)的1.3倍.

圖4 不同質量分數的TA溶液對QAPSF/PVA水凝膠壓縮性能的影響Fig.4 Effect of TA solution with different mass fraction on compression properties of QAPSF/PVA hydrogels
由圖3和圖4可見, QAPSF/PVA水凝膠經適當質量分數TA溶液浸泡處理后, 其拉伸性能和壓縮性能均得到了提高. 其原因可能是當QAPSF/PVA水凝膠浸泡在較低質量分數的TA溶液中時, 少量的TA分子隨水分子進入水凝膠的網絡結構中, TA分子中的羥基可與水分子、TA分子和PVA分子鏈上的羥基形成較強的氫鍵相互作用, 導致QAPSF/PVA水凝膠的拉伸性能和壓縮性能提升. 隨著TA溶液質量分數的增加, 由于TA分子與QAPSF/PVA水凝膠體系形成了較強的氫鍵相互作用, 不僅阻礙了水分子和TA分子進一步向水凝膠網絡滲透, 而且有可能導致水凝膠網絡失水, 因此當TA溶液的質量分數增大到一定程度后, QAPSF/PVA水凝膠的拉伸性能和壓縮性能將隨TA溶液質量分數的增大而下降.
2.4 QAPSF/PVA/TA水凝膠的溶脹性能
圖5為室溫下不同質量分數的TA溶液對QAPSF/PVA水凝膠溶脹性能的影響. 其中, 圖5(A)為浸泡在不同質量分數的TA溶液中QAPSF/PVA水凝膠的溶脹動力學曲線; 圖5(B)為QAPSF/PVA水凝膠浸泡在不同質量分數的TA溶液中, 浸泡時間為24 h時溶脹度的變化曲線. 由圖5(A)可見, 在純水和TA溶液中, QAPSF/PVA水凝膠在達到溶脹平衡前其質量均隨浸泡時間的增加而增加. 在純水中, QAPSF/PVA水凝膠達到溶脹平衡的時間約為16 h; 在TA溶液中, QAPSF/PVA水凝膠達到溶脹平衡的時間約為12 h. 由圖5(B)可見, QAPSF/PVA水凝膠達到溶脹平衡時其平衡溶脹度隨TA溶液質量分數的增加而顯著減少. 其原因可能是當QAPSF/PVA水凝膠浸泡于TA溶液時, TA分子鏈上的羥基與水分子、TA分子和PVA分子鏈上的羥基形成較強的氫鍵相互作用, 導致QAPSF/PVA水凝膠的質量增加. 隨著溶脹時間的增加, TA分子與QAPSF/PVA水凝膠體系形成的氫鍵相互作用阻礙了水分子和TA分子進一步向水凝膠網絡滲透, 導致QAPSF/PVA水凝膠達到溶脹平衡的時間縮短且質量減小.

圖5 不同質量分數的TA溶液對QAPSF/PVA水凝膠溶脹性能的影響Fig.5 Effect of TA solution with different mass fraction on swelling properties of QAPSF/PVA hydrogels
2.5 QAPSF/PVA/TA水凝膠紫外-可見光吸收光譜
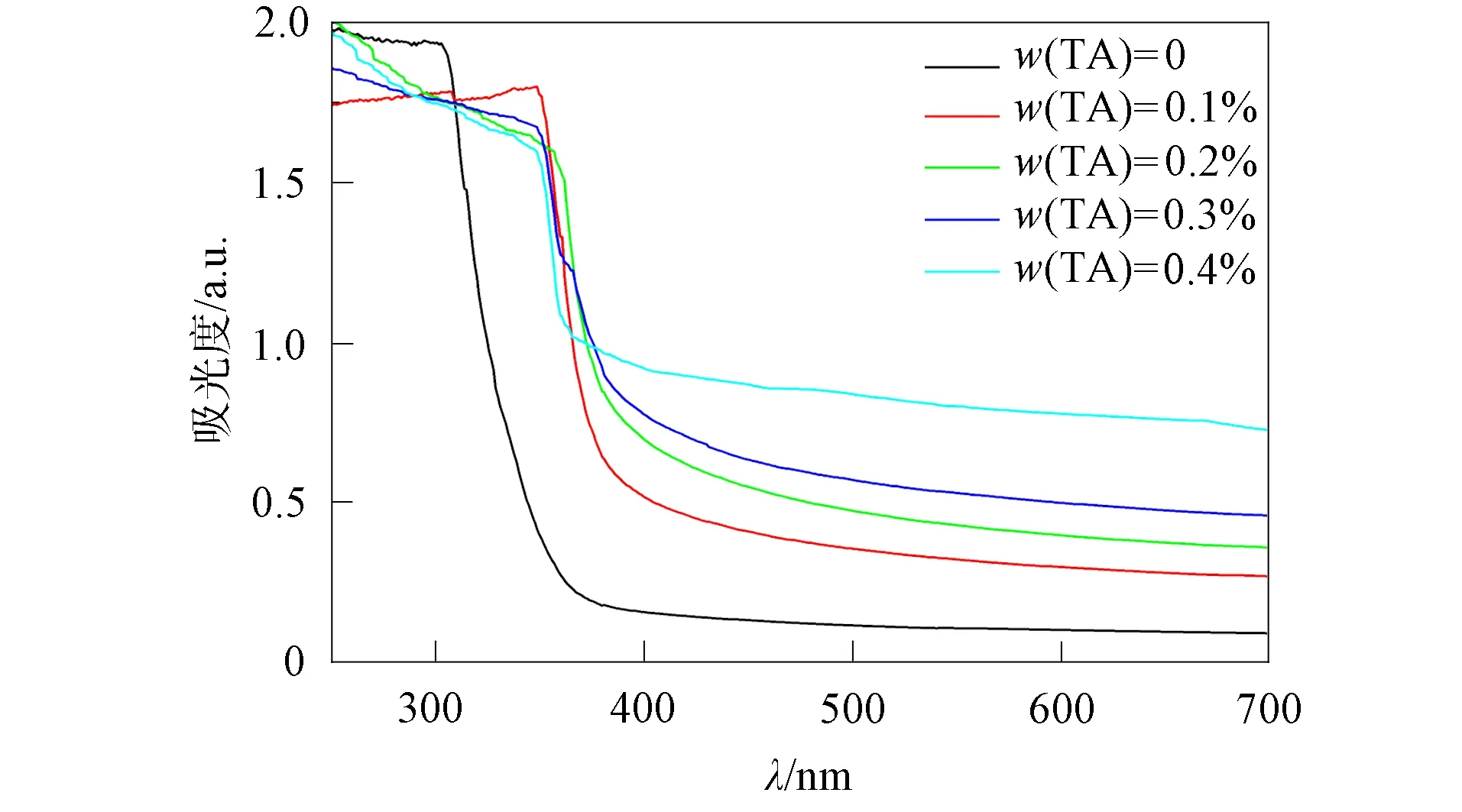
圖6 不同質量分數TA溶液對QAPSF/PVA 水凝膠透明性的影響Fig.6 Effect of TA solution with different mass fraction on transparency of QAPSF/PVA hydrogels
圖6為QAPSF/PVA/TA水凝膠浸泡在不同質量分數TA溶液中的紫外-可見光吸收光譜. 由圖6可見: 在可見光波長400~780 nm內, QAPSF/PVA水凝膠在純水中溶脹后吸光度<0.2, 表明其具有較好的可見光透過率及透明性; QAPSF/PVA水凝膠的吸光度隨TA質量分數的增加而增加, 表明TA可降低QAPSF/PVA水凝膠的透明性. QAPSF/PVA/TA水凝膠在紫外光區的吸光度較高, 具有較強的紫外吸收特性. 在波長為300~360 nm的紅斑效應區內, 浸泡TA溶液后QAPSF/PVA水凝膠的吸光度大于1.5, 表明吸附了TA的QAPSF/PVA水凝膠可作為一種良好的紫外吸收材料.
2.6 QAPSF/PVA和QAPSF/PVA/TA水凝膠的微觀形貌
圖7為QAPSF/PVA和QAPSF/PVA/TA水凝膠的SEM照片, 其中圖7(A)和(B)分別為QAPSF/PVA水凝膠在純水和w(TA)=0.2%溶液中達到溶脹平衡后經冷凍干燥得到的SEM照片. 由圖7可見, QAPSF/PVA水凝膠呈典型的凝膠多孔網絡結構, 這些微孔是由冷凍干燥過程中冰晶升華形成的, 其微孔直徑約為70~90 μm. 與圖7(A)相比, 圖7(B)中水凝膠微孔結構更致密, 平均直徑明顯縮小, 與圖5結果一致, 可見TA降低了QAPSF/PVA水凝膠在溶脹平衡時的吸水率.

圖7 QAPSF/PVA(A)和QAPSF/PVA/TA(B)水凝膠的SEM照片Fig.7 SEM images of QAPSF/PVA (A) and QAPSF/PVA/TA (B) hydrogels
2.7 QAPSF/PVA/TA水凝膠的分子動力學模擬
圖8為QAPSF/PVA/TA水凝膠體系內3種氫鍵相互作用構象圖, 其中: 圖8(A)為PVA分子間氫鍵模型; 圖8(B)和圖8(C)分別為由PVA中羥基作為質子給體, TA中酚羥基和羰基作為質子受體形成的氫鍵模型. 表2為QAPSF/PVA/TA水凝膠體系內3種氫鍵的幾何參數. 根據化學鍵強度理論可知, 鍵長越短鍵角越大的氫鍵, 其鍵能越高. 由圖8和表2可見, PVA與TA之間形成2種氫鍵相互作用的強度均大于PVA分子間形成的氫鍵, 這有利于QAPSF/PVA水凝膠在浸泡TA溶液后其拉伸性能和壓縮性能的提高.

圖8 QAPSF/PVA/TA水凝膠體系內3種氫鍵構象Fig.8 Conformations of 3 types of hydrogen bonds in QAPSF/PVA/TA hydrogel systems

表2 QAPSF/PVA/TA水凝膠中體系內3種氫鍵的幾何參數
2.8 QAPSF/PVA和QAPSF/PVA/TA水凝膠的抑菌性能
QAPSF/PVA復合水凝膠因抑菌官能團位于聚砜高分子鏈上, 由于用抑菌圈法測試抑菌性能時其擴散性較差, 幾乎觀察不到抑菌圈, 因此用GB/T 15979-2002中非溶出性抗(抑)菌產品性能測試方法對其進行測試, 實驗結果列于表3.

表3 QAPSF/PVA和QAPSF/PVA/TA水凝膠的抑菌性能對比
由表3可見: 未浸泡TA的QAPSF/PVA復合水凝膠被試樣品組對大腸桿菌ATCC 25922和金黃色葡萄球菌ATCC 25923的抑菌率與對照樣品組抑菌率的差值分別為47.03%和33.29%, 差值均大于26%, 因此判定該復合水凝膠具有一定的抑菌作用; 浸泡w(TA)=0.2%溶液中的QAPSF/PVA水凝膠, 上述的兩個差值分別為82.25%和71.43%, 表明TA可增加復合水凝膠的力學性能, 并可作為溶出類輔助抑菌劑而增強水凝膠的抑菌性能.
3 結 論
1) 以聚砜為原料制備了DS>1.5的氯甲基化聚砜, 與三甲胺水溶液反應, 經透析、冷凍干燥得到了水溶性的季銨鹽聚砜固體.
2) 季銨鹽聚砜和聚乙烯醇與水混合后, 經γ射線輻射制備了QAPSF/PVA復合水凝膠, 避免了使用具有生物毒性的有機溶劑和其他助劑, 該水凝膠具有一定的抗菌性.
3) 將QAPSF/PVA水凝膠浸泡于w(TA)=0.2%的溶液后, 其拉伸強度和壓縮強度分別提高了4.6倍和1.3倍, TA作為輔助抑菌劑提高了QAPSF/PVA復合水凝膠的抑菌性能, QAPSF/PVA/TA復合水凝膠在300~360 nm紅斑效應區的吸光度大于1.5, 因此可作為一種良好的紫外光吸附材料.
猜你喜歡
中學生數理化·中考版(2022年10期)2022-11-10 09:37:42
中學生數理化·八年級物理人教版(2022年12期)2022-02-14 07:08:42
中學生數理化·八年級物理人教版(2021年12期)2021-12-31 03:23:08
中學生數理化·中考版(2020年10期)2020-11-27 01:59:48
中國生殖健康(2019年2期)2019-08-23 08:12:08
石油化工建設(2018年6期)2018-04-22 03:16:54
產品可靠性報告(2017年7期)2017-09-05 09:49:12
中學生數理化·八年級物理人教版(2017年12期)2017-04-18 12:59:38
汽車觀察(2016年3期)2016-02-28 13:16:26
民生周刊(2014年7期)2014-03-28 01:30:54
