腫瘤細胞利用Notch信號形成促癌細胞因子微環(huán)境的研究進展①
2021-11-26 01:47:52古心怡溫俊杰寧云山南方醫(yī)科大學第一臨床醫(yī)學院廣州510515
中國免疫學雜志 2021年21期
關鍵詞:信號
古心怡 溫俊杰 寧云山③ 李 妍③ (南方醫(yī)科大學第一臨床醫(yī)學院,廣州510515)
Notch 信號是一種高度保守的信號通路,參與細胞生長、分化等過程,由受體(Notch1-4)和配體(Jagged1-2 和Dll1-3-4)組成。配受體結合后,Notch蛋白被水解成胞內段(intracellular domain,ICN)進入細胞核并作用于由RBP-Jκ、MAML1-3 與其他轉錄相關蛋白組成的轉錄復合體CSL,調控下游靶基因如Hes1 等的轉錄。 Notch 信號通路也參與調控腫瘤細胞與免疫細胞的相互作用,影響細胞因子調節(jié)網絡與免疫系統(tǒng)功能,介導腫瘤的發(fā)生發(fā)展。展示了近年來Notch 信號對細胞因子微環(huán)境和免疫系統(tǒng)的影響(表1)。本文將重點探討Notch 信號通路異常激活形成的腫瘤微環(huán)境(tumor microenviron?ment,TME),以及其中各類細胞因子對腫瘤發(fā)生、發(fā)展、衰老、轉移及免疫反應產生的影響。
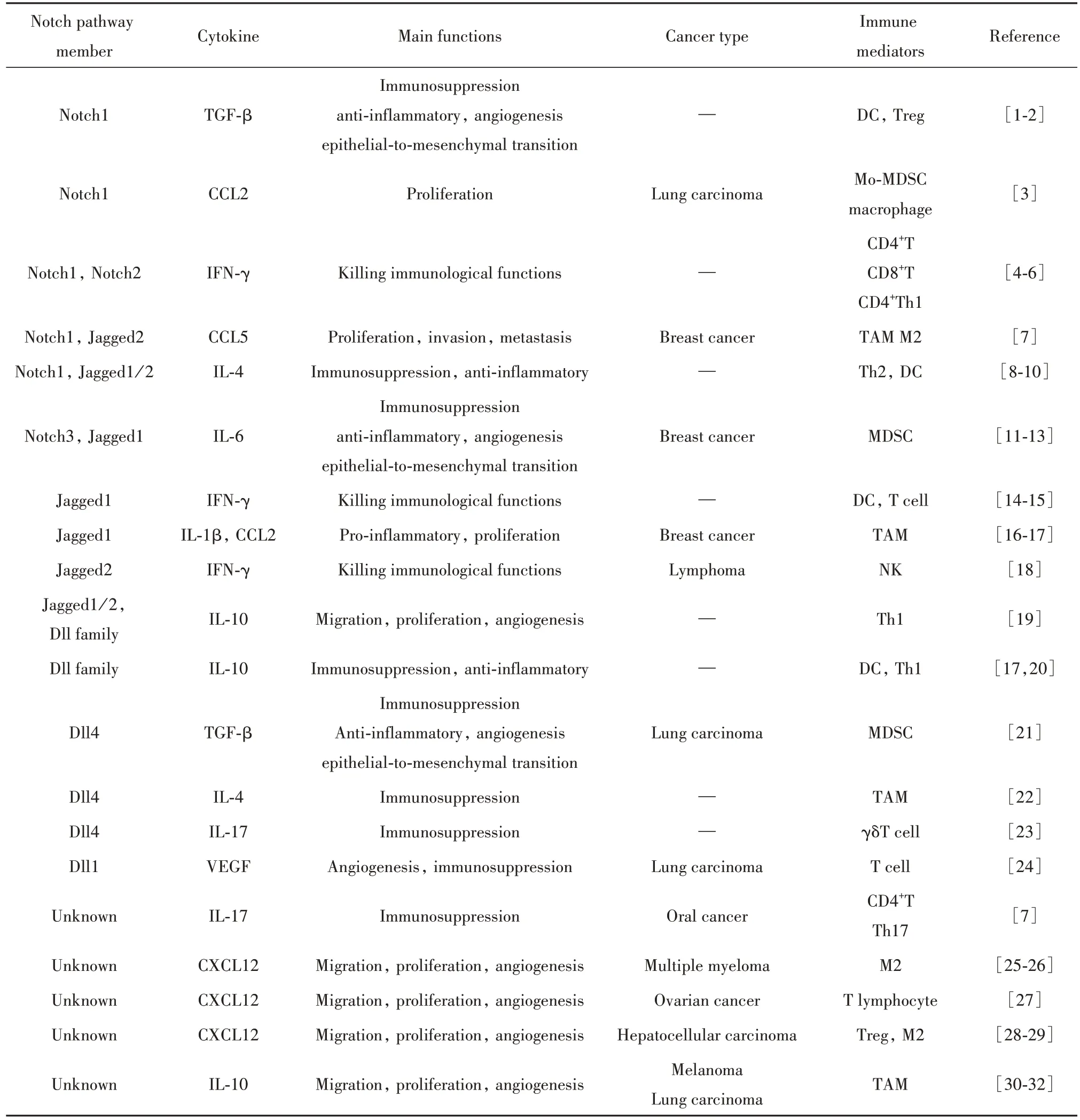
表1 Notch信號在細胞因子微環(huán)境與免疫系統(tǒng)中的作用Tab.1 Effects of Notch signaling on cytokine milieu and immune system
1 Notch 信號通路促進腫瘤相關炎癥細胞因子的分泌并形成免疫抑制微環(huán)境
TME 中存在大量的炎癥細胞因子如TNF-α、IL-1β、IL-4、IL-6、IL-10、IL-17和TGF-β 等,它們構成復雜的細胞因子網絡,調控腫瘤細胞的增殖、侵襲和轉移等,促進腫瘤的發(fā)生發(fā)展[28]。而Notch 信號在其中起到重要的調控作用,并參與免疫抑制微環(huán)境的重構。
1.1 TNF-α TNF-α 是 NF-κB 通路的強效激活因子,促進Notch相關腫瘤疾病的發(fā)生發(fā)展[33]。TNF-α激活IκBα 通路,使轉錄因子FOXA2 磷酸化失活,抑制Notch 阻遏蛋白Numb 的表達,從而解除對Notch的阻遏,并激活該通路[34]。TNF-α 還通過激活IκBβ通路,促進 RBP-Jκ 與 Hes1 基因表達,并抑制抗炎因子PPARγ 的作用,同時募集巨噬細胞與中性粒細胞于癌灶局部釋放 TNF-α、IL-6 與IL-1β,引發(fā)炎癥反應,誘發(fā)炎癥相關腫瘤疾病如肝癌等[34-35]。TNF-α還作為反式作用因子激活MDCS 的p65 通路,上調Jagged1/2,介導MDCS 產生癌抗原耐受,削弱CD8+T細胞殺傷作用[36]。此效應在 Lewis 肺癌、結腸癌、胸腺瘤與黑色素瘤的小鼠腫瘤模型都有典型表現(xiàn)。而在多發(fā)性骨髓瘤(multiple myeloma,MM)中,Notch與TNF-α 相互作用將導致溶骨。MM 細胞與骨細胞共培養(yǎng),二者Notch 信號均會激活,骨細胞會發(fā)生凋亡,而MM 細胞活性增強,分泌大量TNF-α,加快骨細胞凋亡進程。僅下調TNF-α 只能延緩骨細胞凋亡進程,而同時阻斷Notch 信號則能完全逆轉其凋亡,延緩MM發(fā)展[37]。
1.2 IL-1β 促炎因子IL-1β主要由巨噬細胞、中性粒細胞和內皮細胞在急性炎癥反應時分泌,引起機體發(fā)熱與全身性炎癥反應。而腫瘤病灶的IL-1β 主要由癌細胞、TAMs 與脂肪細胞產生,其分泌由Toll樣受體啟動,受Notch 信號調控。IL-1β 具有抗癌與促癌的雙重效應,其偏向性取決于不同的腫瘤環(huán)境[15,38]。Notch 與 IL-1β 相互作用能誘導腫瘤干細胞(cancer stem cells,CSCs)自我更新。如三陰性乳腺癌腦轉移后,癌細胞高水平的IL-1β 會促進鄰近星形膠質細胞高表達Jagged1,激活CSCs 的Notch 信號,促進CSCs 自我更新[39]。在基底樣乳腺癌中,Jagged1 激活的腫瘤細胞釋放ICN1 與ICN3,作用于Notch 基因轉錄起始位點上游 2 085 bp 的 RBP-Jκ 序列,促進IL-1β 分泌,間接提高CCL2 水平,募集腫瘤相關巨噬細胞(tumor associated macrophage,TAM)及單核細胞于腫瘤病灶,形成免疫浸潤,導致不良預后[15,40]。
1.3 TGF-β TME 中間質細胞與 Jagged1 激活的腫瘤細胞均會產生TGF-β,維持微環(huán)境中IL-1β 與CCL2含量,募集單核細胞,形成免疫浸潤,促進腫瘤產生耐藥性并發(fā)生上皮細胞-間質轉化(epithelial-tomesenchymal transition,EMT)。研究表明,Notch 信號能作用于 Th17、Tregs 與 DCs,從而調控 TGF-β 水平。骨腫瘤細胞的Jagged1/2 與Notch1/2 結合激活Notch信號,使ICN入胞結合Smad3蛋白后共同進入細胞核,在RBP-Jκ 的參與下促進Hes 基因表達RANKL,作用于 DCs 和單核細 胞[41]。腫瘤細胞Notch 信號激活還可使其高表達TGF-βR1,提高TGF-β 敏感性[15]。在尿激酶型纖溶酶原激活劑(urokinase-type plasminogen activator,uPA)的作用下,腫瘤相關巨噬細胞(tumor-associated macrophages,TAMs)分泌的高水平TGF-β 結合腫瘤細胞TGF-βR1,會促進腫瘤細胞增殖與腫瘤發(fā)展。綜上,Notch信號調控TGF-β水平,將促進骨腫瘤等其他腫瘤疾病的發(fā)生發(fā)展。
1.4 IL-6 腫瘤細胞與腫瘤間質細胞均能分泌高水平IL-6,并與Notch相互作用,重構機體免疫環(huán)境,提高腫瘤耐藥性、侵襲力與增殖力,導致不良預后[11-12,42-44]。上述效應在 MM 與乳腺癌中最為典型。TME中的腫瘤細胞高表達Jagged1/2與Notch3,前者結合間質細胞受體,后者則結合其他腫瘤細胞配體,導致兩種細胞分泌高水平IL-6,激活非經典Notch 信號,維持腫瘤干性。另外,骨髓來源的抑制性 細 胞(monocyte-myeloid-derived suppressor cell,MDSC)會同時分泌IL-6 與NO,二者均會激活腫瘤細胞 Notch 信號:IL-6 促進 STAT3 與 ICN 結合,正向提高IL-6 水平;而NO 則維持STAT3 水平,起到維持腫瘤細胞干性,使其穩(wěn)定傳代的作用[13]。
1.5 IL-4 IL-4 基因3'端下游的保守區(qū)非編碼序列-2 位中存在兩個 Notch/RBP-Jκ 結合位點,提示Notch信號可直接調控IL-4分泌,并與IL-4存在相互作用[45]。Notch 與 IL-4 相互作用可促進 DCs 成熟與分化:Dll4 激活巨噬細胞Notch 信號后,促進其分泌IL-4,為DCs 成熟與分化提供必要條件。若缺乏IL-4,Jagged1激活DCs后反而抑制其成熟,使幼稚細胞大量聚集[10]。Notch 與 IL-4 相互作用還能促進Th2 分化:DCs 的 Jagged2 激活 T 細胞 Notch,在 IL-4存在下可使其分化為Th2,GATA3 在其中起到關鍵作用,IL-2/IL-2Rα 也參與其中[46]。另外,Notch 信號激活能抑制M2 分化,并使成熟的M1 凋亡,這可能與Hes1蛋白結合STAT3后抑制IL-4R通路有關[22]。
1.6 IL-10 IL-10 由TME 中的腫瘤細胞與免疫細胞如Th1 等分泌,其含量高低將決定腫瘤疾病的嚴重程度。Th1 在IL-12 或IL-27 的作用下,或經DCs的Dll配體激活后,會高表達IL-10,起到抑制自身活性的作用[19]。最近研究表明,Notch-IL-10 調節(jié)軸激活能防止肝炎發(fā)生。肝竇內皮細胞Dll 和Jagged 配體會激活Th1 的Notch 信號,提高Th1 靶基因Hes1與Deltex-1 表達水平,進而促進IL-10 分泌而抑制肝細胞炎癥反應[18]。而 Notch 受體缺陷的 CD4+T 細胞則無法分泌IL-10 防止肝炎發(fā)生。Th1 通過Notch-IL-10 調節(jié)軸促進 TAMs 與 M2 向 M1 分化,成熟后的巨噬細胞分泌 TNF-α 與 IL-12,抑制 IL-10 與 TGF-β分泌,發(fā)揮強大的抗癌作用[30]。用 GSI 和 siRNA 阻斷Notch 或敲除巨噬細胞RBP-Jκ,則會使TAMs 分化為M2,分泌IL-10而抑制Th1活性[32]。
1.7 IL-17 IL-17 主要由Th17 細胞產生,也可由CD8+T 細胞、γδT 細胞、NK 細胞與中性粒細胞產生,主要激活NF-κB 通路,誘導各種促炎因子與趨化因子分泌,募集單核細胞與中性粒細胞,引起局部炎癥反應,這與癌癥的發(fā)生密切相關[47-49]。在不同免疫環(huán)境中,Notch 與IL-17 共同發(fā)揮抗癌作用。病原激活 DCs 的 TLR 通路,上調其 Dll4,激活 Th17 的Notch1,使其表達啟動子Ror-γt,反式激活靶基因表達,促進其分泌更多IL-17[50-51]。而CD4+T細胞Notch激活后會分泌 TGF-β 與 IL-6,促進 Th17 分泌 IL-22與IL-23,從而與IL-17 共同發(fā)揮抗癌效應。Jagged1激活DCs 的Notch 信號后促進其高表達IL-2,促進CD25+分泌 IL-17[52]。Notch 還參與 γδT-17 在胸腺與外周免疫器官內的成熟[23]。IL-6使胸腺上皮細胞高表達 Dll4 配體,激活 γδT-17 的 Hes1 基因表達,促進其胸腺成熟[23];IL-6 還會募集中性粒細胞與單核細胞,促進炎癥反應,發(fā)揮促癌作用[53-55]。另外,腸道與肺內的外周免疫組織細胞也能高表達Dll4 激活γδT-17 的 Notch 信號,促進其外周成熟,并分泌 IL-17和IFN-γ,與TNF-α共同發(fā)揮抗癌作用[7,23,51]。
1.8 CCL5 TME中的CCL5由浸潤性白細胞、間充質干細胞(mesenchymal stem cells)或腫瘤相關成纖維細胞(carcinoma-associated fibroblasts,CAFs)產生,具有促癌作用。MM 的骨髓間充質干細胞和乳腺癌細胞高表達的CCL5 激活TAM 的Notch 信號后使其分化為M2,引發(fā)EMT;還會促進其產生更高水平CCL5,激活癌細胞AMPK 通路,增強糖酵解代謝,產生大量乳酸[56-57]。Notch 信號與 CCL5 的相互作用會導致幾種結果:第一,募集TAM 并促進癌轉移;第二,高濃度乳酸與低濃度葡萄糖影響T細胞代謝,抑制其增殖與活化并誘導其凋亡,同時使浸潤性T 細胞分化為細胞毒性T細胞;第三,對于不依賴葡萄糖代謝的Tregs細胞,其數(shù)目反而增加[58]。
1.9 CXCL12 CXCL12 也稱為間質細胞衍生因子1(stromal-derived factor 1,SDF1),結合 CXCR4 或CXCR7 受體后介導相關趨化因子分泌。血液系統(tǒng)腫瘤如MM 中,Notch 信號激活后,水解的ICN 可作為反式激活因子上調CXCR4 基因表達水平[25]。而CXCR4 基因缺失的鼠造血祖細胞中,ICN1 促進CX?CR4 表達將導致白血病發(fā)生[59]。Notch 與 CXCL12/CXCR4 能夠協(xié)同構造以Tregs 與M2 等細胞為主的免疫抑制型腫瘤微環(huán)境。二者相互作用會募集前體單核細胞,并促進前體單核細胞分化為M2。因而骨髓瘤患者M2的CXCR4水平遠高于正常人[26]。目前已證實,Notch1 在B 細胞慢性淋巴瘤中能直接促進CXCR4 表達,其他血液系統(tǒng)惡性腫瘤如T-ALL 需同時激活CXCR4 通路與Notch1 才能提高CXCR4 水平[60]。Notch 與 CXCL12/CXCR4 的協(xié)同作用在卵巢癌與肝癌等實體瘤中亦有典型表現(xiàn),用DAPT與CX?CL12/CXCR4 抑制劑可阻斷此效應[28]。DAPT 阻斷Notch 后會下調 CXCR4 與 CXCL12 水平,使 Tregs 細胞減少,IFN-γ+/IL-10+CD4+與 CD8+細胞增加,打破免疫抑制型腫瘤微環(huán)境,恢復機體適應性免疫力,增強抗癌能力。
2 Notch信號與IFN-γ相互作用調節(jié)免疫細胞功能
IFN-γ 與 Notch 相互作用在 DCs,CD8+T,CD4+T和 NK 細胞中有明顯表現(xiàn)。Notch 與 IFN-γ 相互作用促進DCs成熟,提高其誘導T 細胞生長分化的能力,發(fā)揮重要的抗癌作用[61]。DCs的Jagged1型Notch 激活后,在CD80/CD86 與PI3K 通路的參與下逐步成熟,分泌 IL-12 與 IL-6,其中 IL-6 為激活多種 T 細胞亞型的必要條件[62]。而 Notch 會阻斷 IFN-γ 對 Th2的誘導作用,促進T 細胞向Th1 分化。MAML-/-轉基因小鼠模型證明,Notch 激活的DCs 能誘導CD4+向 Th1 分化,但若缺乏 IFN-γ,即使 Notch 處于激活狀態(tài),誘導也無法實現(xiàn)[6,63]。這可能由于ICN含量過低,無法充分激活 IFN-γ 基因的 RBP-Jκ 增強子元件[62-63]。因此,T細胞分化為Th1需要IFN-γ與Notch的共同參與。DCs 的 Dll4 或 Jagged1 激活 CD8+T 細胞的Notch2,是CD8+T細胞分裂與活化的前提,也是細胞因子IFN-γ、TNF-α、IL-12、穿孔素和顆粒酶B分泌的條件[4-5]。Notch 對 CD8?DC 與 CD8+T 的作用有所不同。在LPS 的誘導下,前者表達MyD88 依賴性Dll4,激活 Notch 信號,促進 Th1 分泌 IFN-γ。小鼠淋巴瘤模型等相關體內外實驗表明,GM-CSF 與CpG DNA 能夠促進 DCs 表達 Jagged2。另外,DCs 還表達Dll配體,與表達Jagged2和Dll配體的巨噬細胞共同激活 NK 細胞的 Notch2,促進 NK 成熟,分泌 IFN-γ,并增強其細胞毒性,并發(fā)揮重要的抗癌作用[18,64]。
3 Notch 信號與IL-1β、CCL2相互作用介導免疫浸潤
Notch 信號在TME 中會促進腫瘤細胞分泌CCL2。在實驗誘導型肝纖維化與慢急性肝衰竭中,此效應由 Dll4 激活[65];而在其他腫瘤疾病中,則由Jagged1激活[16]。肺癌疾病中,纖維細胞、成骨細胞、內皮細胞和平滑肌細胞均產生高水平CCL2,募集單核-骨髓源性免疫抑制細胞(monocyte-myeloid-de?rived suppressor cells,Mo-MDSCs)與巨噬細胞,促進腫瘤發(fā)展與轉移,而腫瘤抑制因子FBXW7 能降解ICN 與相關癌基因,延緩腫瘤進展,但由于其水平較低,不能充分發(fā)揮作用[3]。腫瘤源性miR-146a 激活骨髓間充質干細胞(bone marrow mesenchymal stem cell,BMSC)Notch1,促進除CCL2 以外的多種細胞因子如IL-8、IL-6、CXCL1、CCL5、IL-10 的分泌,加快腫瘤進展[56]。而黑色素瘤中,沉默Notch 共激動因子MAML1 的黑色素瘤細胞M624 可分泌更高水平CCL2[66]。CCL2會與 Notch 共同促進腫瘤發(fā)展,而肥胖患者有典型表現(xiàn),這與他們的代謝狀況、激素水平和leptin 含量有關[15]。罹患乳腺癌的肥胖患者體內高水平 leptin 可激活Notch、JAK2/STAT、MAPK1/2K 1/2與PI3K/AKT幾種通路,提高IL-1β水平[67-68]。
4 Notch 信號與VEGF 相互作用誘導血管生成與腫瘤浸潤
TME 中腫瘤細胞與間質細胞分泌的VEGF 會誘導血管新生,為腫瘤細胞生長提供氧分與轉移渠道,而Notch 信號在其中起到重要作用。新生毛細血管由前端細胞與莖細胞組成,它們的比例將直接影響新生毛細血管的出芽與成熟。乳腺癌小鼠移植瘤模型中,關鍵分子leptin 與IL-1 共同促進癌細胞分泌VEGF,激活前端細胞VEGFR2/3 與Nrp1 受體后,前端細胞Dll4激活莖細胞Notch1,上調莖細胞VCAM1,有利于中性粒細胞浸潤和腫瘤細胞的血道轉移[67,69]。前端細胞 Dll4 還會促進自身向莖細胞分化,下調前端細胞/莖細胞比例[70]。莖細胞Notch 激活后,下調VEGFR1/2/3 或促進sVEGFR 分泌,中和VEGF,降低環(huán)境中VEGF水平,減緩血管生成。
多發(fā)性骨髓瘤中,BMSC 的Jagged2-Notch 信號激活會促進MM 細胞分泌VEGF,上調前端細胞Dll4,激活莖細胞Notch 信號,促進莖細胞增殖[71-72]。在神經膠質瘤、黑色素瘤、淋巴瘤、纖維肉瘤、胰腺癌、大腸癌、肺癌、乳腺腫瘤小鼠病理模型中,若阻遏Notch信號,則誘導血管內皮高表達VEGFR,增加血管分支密度,降低每條分支的血流灌注量造成缺氧,抑制腫瘤生長。結腸癌小鼠腫瘤模型中,VEGF上調CD8+的PD-1,產生免疫抑制,而VEGF-A-VEG?FR 抗血管生成藥可靶向阻斷此效應[73]。在神經系統(tǒng)腫瘤如多形性膠質母細胞瘤中,癌細胞VEGF-C/VEGFR3 通路與Notch 信號相互作用還能介導癌灶淋巴管的形成,為腫瘤淋巴轉移提供條件[74]。因此,阻斷Notch 信號或VEGF 通路能發(fā)揮靶向抗癌效應。
對于結直腸癌與急性T 淋巴細胞白血病,Dll4則激活腫瘤細胞Notch3,打破腫瘤細胞休眠狀態(tài),使腫瘤細胞重獲增殖力[75]。 目前,人們對Notch 和VEGF 誘導腫瘤血管生成的作用研究較為透徹,但對二者能終止腫瘤休眠的機制卻所知甚少,推測可能與VEGF 和Notch 降低T細胞免疫監(jiān)視功能,打破腫瘤細胞休眠狀態(tài)相關[24]。
5 Notch 信號調控衰老相關細胞因子分泌并介導免疫抑制
Notch 還參與細胞應激反應如DNA 損傷或原癌基因激活,使腫瘤細胞喪失永生化特性與增殖能力而衰老,但仍保留代謝活性與分泌一些細胞因子的能力。衰老的腫瘤細胞仍能募集免疫細胞,發(fā)揮免疫清除功能,有時也會促進毗鄰的非衰老細胞增殖,營造免疫抑制環(huán)境,抑制抗癌作用,加快腫瘤進展。
RBP-Jκ 是 p53 基因的阻遏蛋白,p53 與衰老相關基因p21和促炎因子基因具有相同調控位點。若敲除真皮、口腔粘膜、乳腺和肺組織內原始的腫瘤相關成纖維細胞(CAFs)的RBP-Jk 基因,會導致腫瘤細胞衰老[76]。Notch1,Notch3 分別與 Jagged1 結合后也能導致腫瘤衰老,其中以Notch1 產生的效應最為典型[77]。衰老途徑有多種,但都要經歷兩個特殊復雜的階段。第一階段稱為Notch 誘導型細胞衰老(notch-induced senescence,NIS),分為癌基因誘導型衰老(oncogene-induced senescence,OIS)與DNA損傷型衰老(DNA damage-induced senescence,DDIS)。高水平TGF-β 能抑制CCAAT/增強子結合蛋白β(CCAAT/enhancer-binding protein β,C/EBPβ)轉錄活性,進而抑制促炎細胞因子分泌,影響淋巴細胞黏附與溢出功能,削弱抗癌能力,形成免疫抑制環(huán)境。若ICN1 水平異常提高會使p16INK4A 高表達,p21 和Rb 去磷酸化,細胞周期將永遠停留于G0/G1期,導致細胞停止生長而衰老[78]。進入第二階段,癌細胞完全衰老,Notch 表達則回到基準水平,C/EBPβ 活性恢復,形成衰老相關分泌表型(senes?cence-associated secretory profile,SASP)[77]。綜上,兩種不同的Notch 水平,對應兩種衰老時相細胞因子的分泌:衰老第一階段,以抗炎細胞因子為主,而第二階段,在Notch1與C/EBPβ的相互作用下,則以促炎細胞因子與基質重構酶(MMP1、3、10)為主[77]。
盡管Notch 與其他通路的相互作用導致細胞衰老的機制仍未全部闡明,但其介導衰老相關TME,影響抗癌免疫的效應卻已明晰,這為Notch 信號靶向抗癌療法奠定基礎。
6 結論與展望
細胞因子調節(jié)網絡在腫瘤的發(fā)生發(fā)展中發(fā)揮重要作用,一些信號通路如Notch 信號的異常激活將導致腫瘤細胞與細胞因子調節(jié)網絡發(fā)生異常的相互作用,產生多種復雜的免疫反應,甚至重構患者的免疫環(huán)境[79]。針對這些信號通路的靶向療法理論上可恢復正常免疫環(huán)境,提高治愈率與患者生存率。目前,阻斷Notch 信號的靶向療法大多依賴GSI 但由于缺乏針對性,容易產生胃腸道毒副作用,導致胃黏膜腸上皮化生,因此該應用受到限制。但如今已有Notch1、Notch2/3 和Dll4 單抗藥物與小分子Jagged1/2靶向藥物問世,這些藥物能特異性抑制Notch 信號,減輕副作用[80-84]。但大多數(shù)腫瘤發(fā)病機制并非單純依賴一條信號通路,如Notch 信號會聯(lián)合NF-κB、CXCR4/CXCL、STAT 等其他相關通路發(fā)揮作用,共同導致腫瘤發(fā)生。因此,攻克腫瘤道阻且長,而機制的闡明為抗癌第一關,鑒于Notch 信號對細胞因子網絡的調控作用,未來可以針對其進行抑制,從而干預其介導的腫瘤增殖、侵襲、轉移和血管生成,為腫瘤治療提供潛在的治療靶點。
猜你喜歡
鴨綠江(2021年35期)2021-04-19 12:24:18
考試與評價·高一版(2020年6期)2020-11-02 02:45:24
媽媽寶寶(2019年10期)2019-10-26 02:45:34
中國生殖健康(2019年3期)2019-02-01 06:12:26
鐵道通信信號(2018年11期)2019-01-19 01:15:08
電子制作(2018年11期)2018-08-04 03:25:42
鐵道通信信號(2018年2期)2018-04-18 12:18:10
鐵道通信信號(2016年11期)2016-06-01 12:11:32
鑿巖機械氣動工具(2016年3期)2016-03-01 04:00:25
中國病理生理雜志(2015年8期)2015-12-21 12:38:06
