Ce-MnOx低溫凈化氮氧化物和一氧化碳的催化性能研究
2021-12-15 06:56:36韓新宇劉凱杰邊夢瑤張一波楊向光
無機鹽工業 2021年12期
關鍵詞:催化劑
韓新宇,劉凱杰,邊夢瑤,張一波,3,楊向光,3
(1.中國科學技術大學,安徽合肥230026;2.中國科學院贛江創新研究院;3.中國科學院長春應用化學研究所)
中國積極推進綠色低碳發展,承諾力爭在2030年前實現碳達峰、2060年前實現碳中和[1]。其中,解決工業界排放尾氣凈化問題并構建現代化的綠色工業體系,是碳達峰與碳中和過程中的重點與難點。鋼廠[2]、焦化廠[3]和鋁廠[4]等工廠排出的煙氣溫度通常在200℃以下[5],其中氮氧化物(NOx)和一氧化碳(CO)是煙氣中常見的主要污染成分,因此開發低溫活性強的雙功能協同脫除NOx和CO的催化劑,可實現低溫工業煙氣的深度凈化,對中國完成碳達峰與碳中和的目標具有重要的意義。
煙氣中含有大量的氧氣(O2),尤其是鋼鐵行業低溫燒結煙氣中的O2體積分數達16%,因此本文采用低溫CO催化氧化的方式來處理此類低溫煙氣中的CO[6]。而對于NOx,目前使用最為廣泛的凈化方法是以氨氣(NH3)作為原料,并使用合適的催化劑來進行選擇性催化還原反應,簡稱NH3-SCR反應[7],其反應方程式組如下:
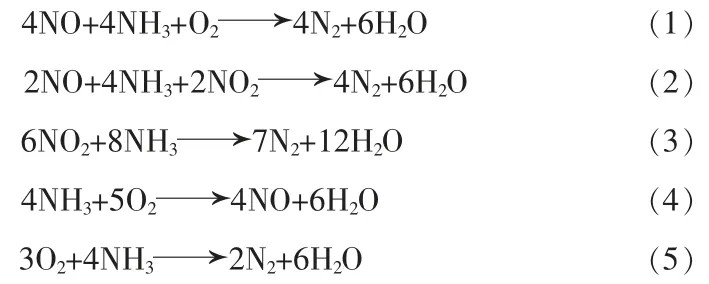
方程式(1)稱為標準SCR反應,適用于一氧化氮(NO)含量多、二氧化氮(NO2)含量少的煙氣。方程式(2)稱為快速SCR反應,反應速率約為標準SCR反應的17倍,其發生率隨著NO2含量增加而增加。方程式(3)為NO2的還原反應,此反應不需要借助O2便可進行,在n(NO2)/n(NO)>1的煙氣中較易發生,但其反應速率遠低于快速SCR反應[8]。方程式(4)為NH3的氧化反應,該反應在高溫下發生,反應中NH3被消耗,產生了NO,進而導致高溫下催化劑的活性急劇下降。方程式(5)為方程式(4)的后續反應,是方程式(1)與方程式(4)的加和,是NH3-SCR反應中的副反應之一,會導致NOx脫除率降低[9];該反應同時也是工業NH3-SCR脫硝過程中處理NH3逃逸的常用手段,即將泄漏的還原劑NH3處理為無污染氣體。
當煙氣溫度高于300℃時,常用的釩-鎢-鈦(V2O5-WO3/TiO2)催化劑表現出優異的催化性能,但對于低溫煙氣而言,V基催化劑在200℃以下無法高效地活化分子氧,進而無法產生更高的催化活性[10]。近年來,研究人員發現錳基氧化物(MnOx)催化劑具有較好的低溫SCR活性,目前已成為低溫脫硝催化劑的研究熱點[11]。
單一的錳氧化物已經具有較好的催化能力,為了進一步優化性能,通常將MnOx與其他元素進行摻雜或修飾。例如,KIM等[12]通過溶膠-凝膠法制備了摻鐵(Fe)的MnO2催化劑,并將其負載至TiO2上,提高了NOx轉化率;ZHANG等[13]制備了物質的量比為1∶1的CuO-MnO2粉體混合物,并將其負載至SAPO-34分子篩上,其NOx轉化率相比摻雜前提高了20%;ZHU等[14]制備了鈷(Co)摻雜的錳氧化物催化劑,形成了Mn2Co1Ox結構,在180℃時NOx轉化率接近100%;ZHANG等[15]向MnOx中同時摻雜了鎂(Mg)和Fe兩種金屬元素,相較于Mn-Fe體系進一步降低了起活溫度。YANG等[16]于1992年發現稀土金屬鈰(Ce)的氧化物(CeO2)本身對于NOx的還原具有一定的催化能力,且CeO2又具有較好的氧儲存性能,有利于O2的吸附活化。因此,本文意圖結合兩種物質的優點,采用Ce來摻雜Mn基催化劑,通過不同方法制備CeMnOx固溶體和CeO2-MnOx無定型混合物,并用于低溫煙氣中NOx和CO的協同脫除。
1 實驗部分
1.1 樣品制備
1.1.1 共沉淀法
正向共沉淀法:將Ce(NO3)3·6H2O固體和質量分數為50%的Mn(NO3)2溶液混合,然后加入48 mL去離子水和12 mL乙醇,此時溶液的離子濃度為0.1 mol/L。隨后向上述溶液中滴加6 mL氨水與54 mL去離子水的混合溶液,靜置并離心去除上清液后,充分洗滌以去除雜質,隨后將沉淀物質在80℃下干燥12 h,并在馬弗爐中在500℃下煅燒4 h。此方法制備的n(Ce)∶n(Mn)=x∶y的樣品記為FC-x∶y。
反向共沉淀法:將上述金屬離子溶液滴加到6 mL氨水與54 mL去離子水的混合溶液中,同上進行后續提純操作,制備了反向共沉淀樣品。此方法制備的n(Ce)∶n(Mn)=x∶y的樣品記為RC-x∶y。
1.1.2 球磨法
將CeCl3·7H2O固體與過量NaOH固體混合,加入兩倍于所有固體原料質量的磨球,在球磨機中以540 r/min的速度球磨1 h,以創造良好的堿性環境。再向球磨罐中加入KMnO4固體,球磨1 h后,將磨球取出;對產物進行充分洗滌、離心并在80℃下干燥12 h。此方法制備的n(Ce)∶n(Mn)=x∶y的樣品記為BM-x∶y。
1.1.3 溶液燃燒法
將Ce(NO3)3·6H2O固體和質量分數為50%的Mn(NO3)2溶液混合,加入聚乙二醇(PEG),再加入10 mL去離子水和5 mL乙醇。攪拌至固體完全溶解后,在70℃下烘干至溶液為膠狀,后放入馬弗爐中,在500℃下煅燒4 h。此方法制備的n(Ce)∶n(Mn)=x∶y的樣品記為SC-x∶y。
1.1.4 溶膠-凝膠法
將Ce(NO3)3·6H2O固體和質量分數為50%的Mn(NO3)2溶液混合,加入60 mL去離子水和0.012 mol一水檸檬酸,然后用氨水將pH調節至8.0。在70℃下水浴并攪拌至凝膠出現,于80℃下干燥12 h后,在馬弗爐中在500℃下煅燒4 h。此方法制備的n(Ce)∶n(Mn)=x∶y的樣品記為SG-x∶y。
1.2 測試與表征
1.2.1 催化劑性能測試
CO與NOx單獨催化性能測試:將制備得到的催化劑分別置于兩臺固定床反應器中,并采用GC2060氣相色譜儀和Hiden HPR-20EGA質譜儀分別對CO和NOx的脫除率進行檢測分析。其中,CO催化氧化測試所用的氣體中含有體積分數為1%的CO和99%的空氣,測試空速為60 000 mL/(g·h);在NH3-SCR測試中使用的氣體中含有O2、NH3、NO和氬氣(Ar),它們的體積分數分別為5%、0.05%、0.05%和94.9%,測試空速為100 000 mL/(g·h)。
CO與NOx協同催化性能測試:將組成(含量以體積分數計)分別為NO(0.05%)、NH3(0.05%)、CO(0.25%)、O2(5%)、N2(19.5%)和Ar(75.15%)的氣體通入固定床反應器,空速為100 000 mL/(g·h),檢測CO和NOx的脫除率。
催化劑持續性能測試:將組成(含量以體積分數計)分別為NO(0.05%)、NH3(0.05%)、CO(0.25%)、O2(5%)、N2(19.5%)和Ar(75.15%)的氣體通入固定床反應器,空速為100 000 mL/(g·h),在200℃下保持20 h,并持續監測CO和NOx的脫除率。
1.2.2 催化劑的表征
使用D8 Advance X射線衍射儀對所制備的樣品進行X射線粉末衍射分析(XRD),以表征其物相組成;使用AutochemⅡ2920化學吸附儀對樣品進行氨氣程序升溫脫附測試(NH3-TPD)和氫氣程序升溫還原測試(H2-TPR),同時使用ASAP2460物理吸附儀進行氮氣吸附-脫附等溫線測試,以檢測樣品的比表面積。
2 結果與討論
2.1 CO催化氧化性能
圖1是采用不同方法制備的CeMn氧化物催化劑的CO催化氧化活性與溫度的關系圖。其中,圖1a是采用共沉淀法制備的3種不同比例的樣品的CO脫除率與溫度曲線圖。由圖1a可知,所有催化劑起活溫度均低于100℃,且在160℃時都能夠達到100%的CO轉化率。反向共沉淀法制備的催化劑整體性能優于正向共沉淀法制備的催化劑。反向共沉淀法制備的樣品中RC-2∶1活性最高,在120℃時達到了100%的CO轉化率,整體趨勢為Ce元素占比越高,催化性能越強;而正向共沉淀法所制備的催化劑相反,Ce元素占比越高,催化活性越低,且FC-2∶1樣品活性最低,其原因可能是溶液體系中堿性環境的強弱導致催化性能存在差異。金屬離子溶液為酸性,其pH約為3,向其中加入氨水,環境由酸性逐漸轉變為堿性。由于Ce和Mn的溶度積差異[Mn(OH)2,KSP=1.9×10-13;Ce(OH)3,KSP=1.5×10-20]導致Ce優先沉淀,但由于溶液pH較低,Mn不能和Ce同時沉淀,不利于CeMnOx固溶體的形成。而反向共沉淀法是將pH低的金屬離子溶液加入pH高于11的氨水溶液中,Ce和Mn在高堿性環境下能夠同時沉淀,CeMnOx一經形成便不再變化,因此結構完整,催化效率較高。
圖1b是采用球磨法制備的3種不同比例的樣品的CO脫除率與溫度的關系曲線圖。由圖1b可知,在180℃以下時BM-1∶2催化劑性能最好,且在160℃時能達到80%以上的CO轉化率;在180℃以上時,3種比例的催化劑性能接近,都能夠在200℃時將CO全部轉化為CO2。
圖1c是采用溶液燃燒法制備的3種不同比例的樣品的CO脫除率與溫度的關系曲線圖。由圖1c可知,3種不同比例的催化劑性能差異明顯。在相同溫度下SC-1∶2具有最高的CO轉化效率,在120℃時CO轉化率達到了100%;其次為SC-1∶1催化劑,在140℃時將CO完全轉化。而SC-2∶1的性能不佳,在180℃時達到100%的CO轉化率。由以上結果可知,溶液燃燒法制備的催化劑中Mn元素占比越高,催化性能越強。
圖1d是溶膠-凝膠法制備的3種不同比例的樣品的CO脫除率與溫度的關系曲線圖。由圖1d可知,與溶液燃燒法不同,SG-1∶1具有最優異的CO催化氧化性能,在140℃時達到了100%的CO脫除率,并在后續的升溫過程中保持不變;其次是SG-1∶2和SG-2∶1,分別在160℃和180℃達到了100%的CO轉化率。SG-1∶1樣品具有最佳的CO轉化性能,因為在使用檸檬酸形成溶膠后,再用氨水將檸檬酸-金屬離子絡合物溶液的pH調節至8.0,隨后在pH=8.0的環境和70℃的蒸發溫度下,CeMn-檸檬酸絡合物膠體粒子長大和膠體體積收縮同時進行[17],最終在形成n(Ce)∶n(Mn)=1∶1的凝膠時可以最大限度地增加固溶體的缺陷空間[18],因此表現出了最優的表面特性,即最優的催化性能。
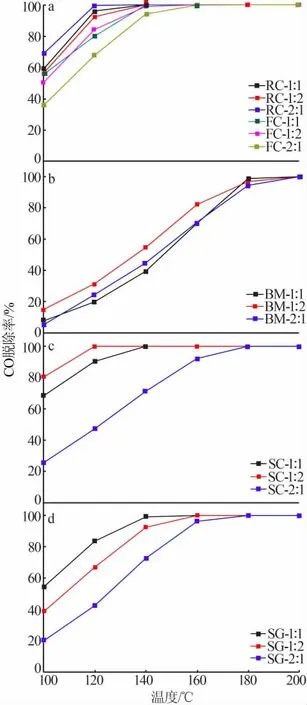
圖1 不同方法制備的鈰摻雜錳氧化物催化劑的CO脫除率-溫度曲線Fig.1 CO removal rate-temperature curves of cerium-doped manganese oxide catalyst prepared by different methods
綜合對比以上4種制備方法所得到的樣品性能,可見共沉淀法制備的催化劑性能最優,主要表現在起活溫度低、低溫下轉化率高和無高溫衰減3個方面;其次是溶液燃燒法所制備的樣品,其性能與共沉淀法制備的樣品相似,但不同比例的催化劑之間性能相差較大。對于溶膠-凝膠法所制備的樣品,其催化能力隨溫度上升而快速上升,但由于起活溫度較高,在160℃以上才能夠轉化98%以上的CO。最后,球磨法制備的樣品催化氧化能力較低,在180℃時尚未完全將CO轉化為CO2。
2.2 NOx催化還原性能
在固定床反應器上測試了催化劑的NH3-SCR性能,并用質譜儀進行檢測,結果繪制成NOx脫除率-溫度曲線,如圖2所示。
圖2a記錄了采用共沉淀法制備樣品的性能數據。由圖2a可知,通過反向共沉淀法制備的3種催化劑性能相近,在100℃時NOx的轉化率已經接近90%,其性能隨溫度上升而略有上升后保持不變。正向共沉淀法制備的樣品性能差異較大,FC-1∶1催化劑性能優于所有反向共沉淀法制備的樣品,位于所有參與測試樣品的首位;FC-1∶2和FC-2∶1性能在160℃以下與其他樣品有較大的差距,其活性隨溫度上升而大幅上升后保持穩定。這說明在FC-1∶1的催化劑中出現了較為顯著的CeMn協同作用。
圖2b呈現了由球磨法制備的3種不同比例的樣品的NOx脫除率與溫度的關系曲線圖。由圖2b可知,與CO催化氧化性能的測試結果不同,在140℃以上BM-1∶1催化劑性能明顯優于其他2份樣品,且該催化劑的性能隨溫度的上升有較大幅度的提升。其余兩種催化劑的NOx脫除率隨溫度變化的趨勢一致,整體性能均落后于BM-1∶1。3種催化劑之間出現了非常明顯的性能差距,原因是BM-1∶1中出現了Ce與Mn兩種元素的協同作用。
圖2c是溶液燃燒法制備的3種不同比例的樣品的NOx脫除率與溫度的關系曲線圖。由圖2c可知,3種催化劑NOx脫除率隨溫度變化的趨勢一致。在120℃以上SC-1∶1和SC-1∶2的活性十分接近,遠高于SC-2∶1催化劑。此現象表明,在測試條件下SC-2∶1無法提供足夠的活性位點,導致大量氣體無法吸附在催化劑表面,降低了催化性能。
圖2d是溶膠-凝膠法制備的3種不同比例的樣品的NOx脫除率與溫度的曲線關系圖。由圖2d可知,3種催化劑的起活溫度不同,催化能力隨溫度上升而上升的幅度也不同。SG-2∶1催化劑在100℃時的性能與其他兩種比例的樣品差距較大,但其催化能力隨溫度升高迅速增加,在140℃以上時其性能已經與SG-1∶1相同。這種現象可能是由于Mn進入CeO2的晶格后,Ce原子在晶格表面覆蓋率高于Mn原子,導致活性組分與氣體無法充分地接觸,無法使更多NOx參與反應。

圖2 不同方法制備的鈰摻雜錳氧化物催化劑的NOx脫除率-溫度曲線Fig.2 NOx removal rate-temperature curves of cerium-doped manganese oxide catalyst prepared by different methods
對比以上4種制備方法制得的催化劑的性能,可以發現共沉淀法制備的樣品起活溫度最低,且同溫度下性能最強;其次是溶膠-凝膠法制備的催化劑,在140~180℃轉化率平穩。溶液燃燒法制備的樣品測試所得曲線與溶膠-凝膠法相似,但整體性能均低于溶膠-凝膠法制備的樣品。最后,球磨法制備的樣品催化效率隨溫度變化的波動較大,且其效率在低溫下不穩定。
2.3 CO與NOx協同催化性能與持續性能測試
由于共沉淀法制備的催化劑在2.1節和2.2節的CO催化氧化性能測試與NOx催化還原性能測試中均位于第一位,且相對于其他3種方法制備的催化劑具有較大幅度的性能優勢,因此選擇RC-1∶1和FC-1∶1進行CO與NOx協同催化性能測試,將測試結果分別繪制為CO脫除率-溫度曲線和NOx脫除率-溫度曲線,如圖3a所示。
由圖3a可知,兩種催化劑在180℃以上的性能相同,都能夠將98%以上的CO轉化為CO2。在低溫區段,RC-1∶1性能優于FC-1∶1,在140℃達到了85%的CO轉化率,而FC-1∶1在此溫度下的CO轉化率為80%。對于NH3-SCR反應,FC-1∶1和RC-1∶1催化劑在測試的溫度區間內性能較為穩定,且在所測試的溫度區間內均能脫除80%以上的NOx。
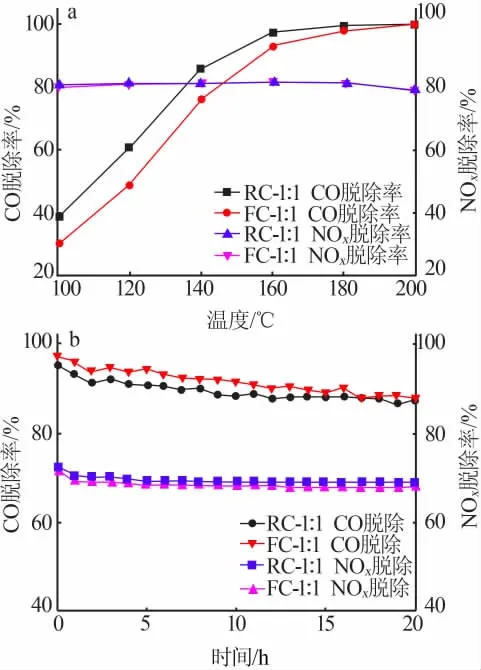
圖3 催化劑在協同催化工況下的CO脫除率-溫度曲線與NOx脫除率-溫度曲線(a);催化劑在協同催化工況下運行20 h的CO脫除率-溫度曲線與NOx脫除率-時間曲線(b)Fig.3 CO removal rate-temperature curves and NOx removal rate-temperature curves of catalysts in synergistic catalysis reaction(a);CO removal rate-time curves and NOx removal rate-time curves of catalysts for 20 h in synergistic catalysis reaction(b)
在協同催化反應中,由于活性位點出現了競爭吸附的現象,催化劑有了較為明顯的性能下降,以至于FC-1∶1和RC-1∶1均不能在較低的溫度達到100%的CO轉化率,相比2.1節的CO催化氧化性能測試有較大幅度的性能降低;而NH3-SCR性能相較于2.2節的NOx催化還原性能測試下降約10%,但其性能隨溫度變化的趨勢仍然保持不變。
將兩種催化劑在固定床反應器上加熱至200℃,并通入混合氣,進行時長20 h的協同催化持續性能測試,并將實驗實時結果繪制為CO脫除率-時間曲線和NOx脫除率-時間曲線,如圖3b所示。實驗誤差原因導致持續性測試開始時的活性略低于圖3a中活性。兩種催化劑在持續運行1 h后均出現了小幅度的性能衰減,CO脫除率從接近100%降低至90%,NOx脫除率從75%降低至70%,隨后保持不變。該測試證明共沉淀法制備的催化劑不但具有優異的催化性能,且能夠在20 h的長時間使用下保持較高活性和穩定性。
為進一步探究催化劑在長時間運行前后的結構變化,使用X射線衍射對兩種樣品的晶體結構進行了分析,如圖4所示。由圖4可以看出,RC-1∶1(圖4a)和FC-1∶1(圖4b)在進行持續測試前與標準CeO2卡片吻合良好,曲線較為平滑,表明結晶度較好;在進行20 h的持續性能測試后,兩種催化劑的XRD譜圖均出現了低強度雜峰,表明在催化反應的過程中,晶格中的O原子在離開后,混合氣中的O2并未完全進入催化劑晶格以填補空位,導致晶體的部分晶面出現缺陷,在X射線衍射中以低強度峰的方式呈現。同時,40~60°衍射角內的峰出現了強度降低的現象,證明(221)面、(311)面和(222)面是參與反應的主要晶面,Ce原子和Mn原子在此晶面分布較多,在長時間測試后結晶度有所下降。

圖4 催化劑進行穩定性測試前后的XRD譜圖Fig.4 XRD patterns of the catalysts before and after the stability test
2.4 XRD分析
選擇RC-1∶1、BM-1∶1、SC-1∶2和SG-1∶2這4種催化劑作為不同方法制備的典型樣品進行了X射線衍射分析,結果如圖5所示。由圖5可知,溶液燃燒法和共沉淀法所制備的樣品與標準CeO2晶型卡片吻合良好,同時衍射峰出現右移,證明Mn原子進入CeO2的晶格之中,使其晶格參數發生變化,即生成了CeMnOx固溶體;溶膠-凝膠法和球磨法制備的催化劑則以CeO2和MnOx粉體混合物為主,結晶度較差,故衍射峰不高,且沒有明顯的晶體結構。
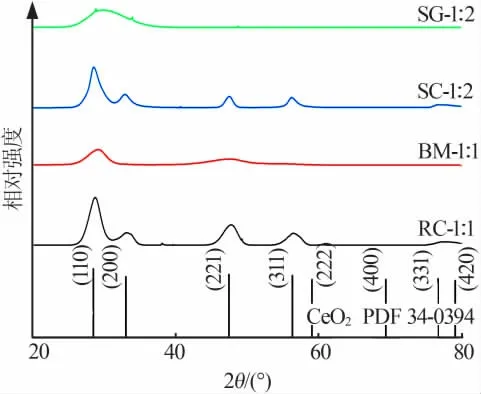
圖5 不同制備方法得到的典型樣品的XRD譜圖Fig.5 XRD patterns of the catalysts obtained by different preparation methods
2.5 NH3-TPD分析
選擇2.4節中的4種催化劑進行NH3-TPD測試,結果如圖6所示。根據TPD測試的原理,脫附峰出現時間先后與酸性的相對強弱成反比關系[19],由圖6可知,當脫附溫度在300℃以下時,共沉淀法和球磨法制備的樣品吸附的NH3首先解吸,對應溫度約為100℃;隨后溶膠-凝膠法和溶液燃燒法制備的催化劑在170℃時解吸,表明這4種樣品中均含有一定量的弱酸位,結合NH3-TPD實驗結果可知弱酸性位點含量并不是影響催化活性的決定性因素。
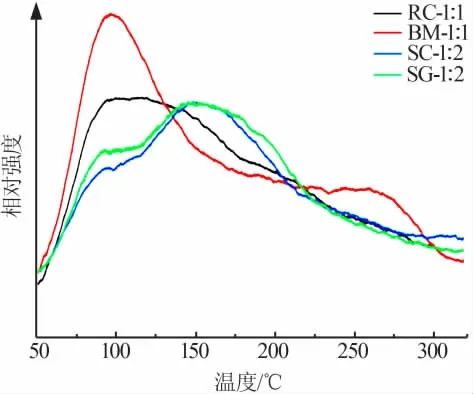
圖6 催化劑的NH3-TPD曲線Fig.6 NH3-TPD curves of catalysts
2.6 H2-TPR分析
選擇2.4節中的4種催化劑,進行了H2-TPR測試,并將測試結果繪制為相對強度-溫度曲線,結果如圖7所示。由圖7可知,采用共沉淀法、溶液燃燒法和溶膠-凝膠法制備的樣品中含有多個還原峰,原因是CeMnOx中MnOx在還原時按照MnO2、Mn2O3、Mn3O4的順序依次進行,其中MnO2在眾多錳氧化物中具有最高的催化活性[20]。RC-1∶1、SC-1∶2和SG-1∶2催化劑中含有較大量的MnO2,因而表現出卓越的NH3-SCR催化能力;球磨法制備的樣品中Mn元素并不是單純以MnO2的形式存在,且具有高催化活性的錳氧化物含量也是四者中最少的,因此在還原過程中400℃以下的峰面積最小,催化性能較差。
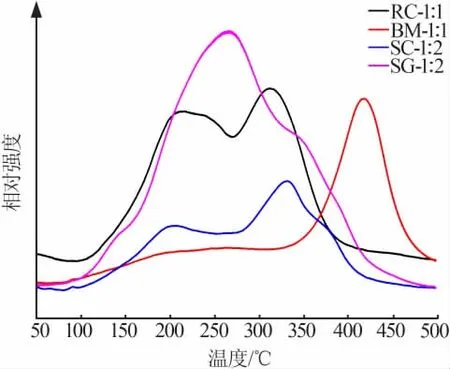
圖7 催化劑的H2-TPR曲線Fig.7 H2-TPR curves of catalysts
2.7 氮氣吸附-脫附測試
選擇2.4節中的4種催化劑,采用氮氣吸附-脫附等溫線測定樣品的BET比表面積,結果如表1所示。由表1可知,共沉淀法制備的樣品比表面積最大,其次是溶液燃燒法制備的樣品;球磨法和溶膠-凝膠法制備的樣品比表面積相近,都小于50 m2/g。在CO氧化性能測試中,催化劑性能的強弱與比表面積大小呈正相關;但在NH3-SCR測試中,由于不同制備方法所得到的氧化物形成機制不同,且NH3-SCR與CO氧化的機理完全不同,溶膠-凝膠法制備的樣品雖然比表面積較小,但其性能在NH3-SCR中排在第二位。因此,對于CO催化氧化反應,比表面積大小能夠影響催化劑的活性;對于NH3-SCR反應,比表面積的大小與氣體吸附位點的多少相關,但由于不同制備方法得到的催化劑所含的活性位點數量不同,且存在CeMn兩種元素的相互作用,因此減弱了比表面積對催化劑性能的影響。
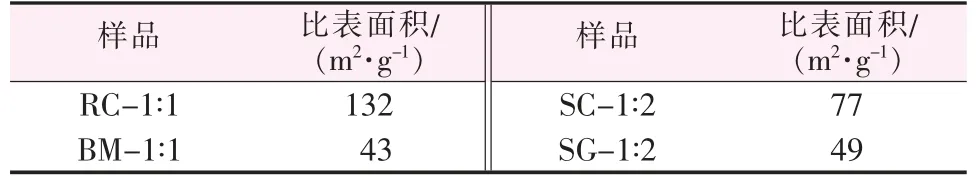
表1 催化劑的比表面積Table 1 Specific surface area of catalysts
3 結論
1)通過不同的制備方法(共沉淀法、球磨法、溶液燃燒法和溶膠-凝膠法)成功合成了4種不同的CeMnOx/CeO2-MnOx催化劑,并對其進行了CO氧化和NOx選擇性還原的催化性能測試。活性測試結果顯示,不同的制備方法對催化劑的催化性能有很大的影響。其中,共沉淀法制備的催化劑整體表現最為優異,在單獨的CO催化氧化性能測試和NOx催化還原性能測試中,140℃時該催化劑的CO轉化率大于90%,NOx轉化率大于80%,且性能隨著溫度上升而繼續增加;在協同催化測試中,該類催化劑出現了不同程度的性能降低,但仍然能夠在200℃時轉化全部的CO和80%以上的NOx。經過20 h的持續運行,該類催化劑出現了小幅度的活性衰減,但仍然能夠脫除70%以上的NOx和80%以上的CO。
2)通過多種表征技術對催化劑的物化性質和結構組成進行了探討,結果表明不同的制備方法對催化劑的結構和性質產生了不同程度的影響。不同方法制備的催化劑在XRD測試中結果差異較大,其中共沉淀法制備的樣品最符合標準CeO2晶型;在化學吸附測試中,球磨法制備的樣品具有最多的弱酸位點,而溶膠-凝膠法制備的樣品具有最多的氧化性位點。共沉淀法制備的催化劑具有最高的比表面積,能夠暴露更多的活性位點,從而表現出較高的CO催化氧化和NOx選擇性還原脫除能力。
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
