西維來司他對急性肺損傷小鼠NLRP3炎癥小體的作用研究*
2021-12-27 06:31:48韓艷奇高志丹陳清杰黃翠萍
湖北科技學院學報(醫學版) 2021年5期
關鍵詞:小鼠
韓艷奇,高志丹,陳清杰,黃翠萍
(1.湖北科技學院藥學院,湖北 咸寧 437100;2.湖北科技學院糖尿病心腦血管湖北省重點實驗室;3.湖北科技學院護理學院)
西維來司他(sivelestat,SV)是一種選擇性中性粒細胞彈性蛋白酶抑制劑,已被報道對內毒素誘導的倉鼠、豚鼠和綿羊肺損傷有效[3-4]。在日本進行的Ⅲ期和Ⅳ期研究顯示[5-6],SV能縮短患者機械通氣時間和重癥監護病房的停留時間,延長急性肺損傷患者的生存期。但SV與NLRP3炎癥小體的關系研究甚少,本研究通過脂多糖LPS復制急性肺損傷小鼠模型,檢測肺組織NLRP3炎癥小體及其相關信號分子的表達變化,探討SV對急性肺損傷小鼠肺組織的作用和機制。
1 材料與方法
1.1 試劑與儀器
1.1.1 主要試劑與動物
32只7周齡雄性C57BL/6小鼠,體質量為22~26g,購自遼寧長生生物技術股份有限公司。LPS購自北京索萊寶科技有限公司(L8880-10mg);OLT 1177購自上海易匯科技有限公司(HY-W011082);SV購自上海易匯科技有限公司(HY-17443);ELISA試劑盒購自武漢赫爾生物科技有限公司(SU-BN20174);NLRP3抗體購自CST(15101S);CASPASE-1購自ABclonal(A0964);p-NF-κB抗體購自ABclonal(AP0123);NE(ELANE)購自ABclonal(A13015);IL-18購自Immunoway(YN1926);IL-1β購自Immunoway(YT2322);β-actin抗體購自ABclonal(AC026);BCA蛋白定量試劑盒,購自上海翊圣生物科技有限公司。
1.1.2 儀器
水平搖床(海門市其林貝爾儀器制造有限公司);電泳槽、電轉膜儀(Bio-Rad公司);多功能酶標儀(美國Bio-Tek 公司);化學發光凝膠成像系統(Bio-Rad公司);研磨儀(武漢塞維爾生物科技有限公司)。
1.2 方法
1.2.1 動物模型與實驗分組
將32只C57BL/6小鼠在標準動物飼養間內,給予無菌水和食物飼養一周,隨機分為4組(每組8只):正常對照組(CON)、模型組(LPS)、模型+SV組(LPS+SV)、模型+NLRP3抑制劑組(LPS+OLT)。正常對照組腹腔注射生理鹽水(20mg/kg);模型組腹腔注射LPS (20mg/kg),制備急性肺損傷(ALI)模型;SV治療組在造模前5d每天腹腔注射SV(100mg/kg);NLRP3抑制劑組在造模前12h內腹腔注射OLT 1177(200mg/kg),分5次給藥,最后一次腹腔注射OLT 1177(40mg/kg)1h后,腹腔注射LPS(20mg/kg)。4組小鼠均12h后處死,留取標本,進行下一步實驗。
2.1 開發主體環保意識薄弱 旅游資源作為一種資源,不論是可再生還是不可再生,在其使用過程中不可避免會受到破壞,絕大多數生態旅游資源都具有破壞容易恢復難的特性。隨著景區知名度的提升,加上游客日益多樣化的需求,旅游目的地不得不想辦法對資源進行深度的挖掘和再次開發。受經濟利益的驅使,常常使得旅游開發具有盲目性,可能會使得資源過度開發而超過自身恢復限度。張家界國家森林公園,由于對修建性建設項目控制不夠,造成水質污染,曾被聯合國教科文組織亮出黃牌警告,當地政府為了恢復景區原貌,花費了上億元人民幣。
1.2.2 肺含水量測定
取右肺上葉,吸水紙吸干表面水分及血液,稱重。置于80℃烘箱72h后稱量肺干重,計算濕干重之比(W/D)。
1.2.3 小鼠肺組織中IL-1β表達水平
小鼠頸椎脫臼處死,取右肺下葉部分組織,稱重,加入PBS緩沖液(組織∶PBS=1∶9),冰上剪碎組織,采用研磨儀充分勻漿,收集上清。通過酶聯免疫法檢測肺組織中IL-1β的表達水平,操作均按ELISA試劑盒說明書進行。
1.2.4 小鼠肺組織中NLRP3炎癥小體相關蛋白以及中性粒細胞彈性蛋白酶表達水平
取適量肺組織,稱重,加入一定體積比例(組織∶RIPA=1∶30)的RIPA裂解液、PMSF、磷酸酶抑制劑A液、磷酸酶抑制劑B液混合液(RIPA∶PMSF∶PA∶PB=100∶1∶1∶1),在碎冰上剪碎組織,冰上靜置20min,裂解小鼠肺組織,利用研磨儀勻漿,離心,取上清,酶標儀562nm波長條件下進行蛋白定量,蛋白含量采用BCA試劑盒檢測。定量后的蛋白樣品取10μL進行上樣,采用8%~15%的SDS-PAGE分離膠進行蛋白電泳,PVDF膜轉膜2h,室溫下用5%脫脂奶粉進行封閉1h,用ECL化學發光顯影,凝膠成像系統檢測NLRP3、p-NF-κB、CASPASE-1、IL-1β、IL-18、NE蛋白的表達變化。
1.2.5 肺組織病理損傷觀察
取各組小鼠右肺中葉組織,迅速放入4%多聚甲醛液中固定,脫水,石蠟包埋,切片,然后用蘇木精-伊紅(hematoxylin-eosin,HE)染色,光學顯微鏡下觀察。
1.3 統計學方法
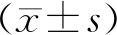
2 結 果
2.1 各組小鼠臨床表現
CON組小鼠活動狀態正常,正常攝食、正常飲水。模型組在腹腔注射LPS 12h時,出現步態蹣跚、精神萎靡、呼吸淺快或深快、節律不齊,時有呼吸困難、眼神渙散。小鼠均存活,無死亡。
2.2 肺含水量比較
與CON組相比,LPS組小鼠肺W/D升高,差異有統計學意義(P<0.05);與LPS組比較,LPS+SV組顯著下降,差異有統計學意義(P<0.05);LPS+OLT組與LPS組比較有所下降,但無統計學差異(P>0.05)。見圖1。

與CON組比較,*P<0.05;與LPS組比較,#P<0.05
2.3 肺組織中炎癥因子IL-1β含量的比較
ELISA法測定結果顯示,與CON組比較,LPS組IL-1β表達水平顯著升高(P<0.05);LPS+SV組IL-1β表達水平較LPS組高,而LPS+OLT組IL-1β表達水平較LPS組低(P<0.05),作用與CON組相當(見圖2),此結果表明,SV不能夠抑制NLRP3炎癥小體下游的炎癥因子IL-1β的釋放。
與CON組比較,*P<0.05;與LPS組比較,#P<0.05
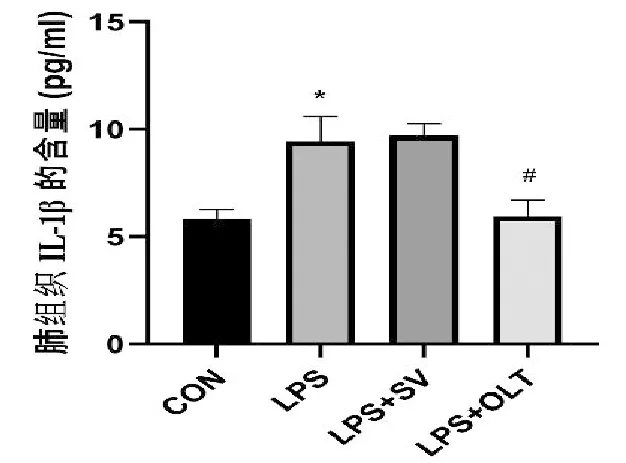
2.4 SV對NE蛋白的影響
如圖3所示,Western blotting結果顯示:與CON組相比,LPS組中性粒細胞彈性蛋白酶(NE)表達顯著性增多(P<0.05);SV作為一種選擇性中性粒細胞彈性蛋白酶抑制劑,與LPS組相比較,能夠顯著性抑制NE的表達(P<0.05),且治療作用與CON組相當;而NLRP3炎癥小體抑制劑并沒有降低NE的表達。
與CON組比較,*P<0.05;與LPS組比較,#P<0.05
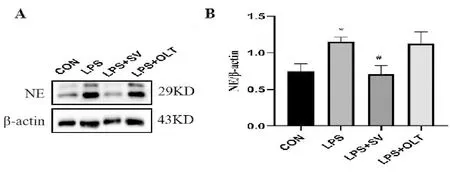
2.5 SV對p-NF-κB蛋白的影響
Western blotting法檢測各組小鼠肺組織中的p-NF-κB蛋白表達變化,結果見圖4。與CON組比較,LPS組p-NF-κB蛋白表達明顯增多(P<0.05);與LPS組相比,LPS+SV組p-NF-κB蛋白表達明顯減少(P<0.05);同時,OLT 1177作為NLRP3炎癥小體特異性抑制劑,抑制了p-NF-κB蛋白表達,但統計學上無意義(P>0.05)。
與CON組比較,**P<0.01;與LPS組比較,##P<0.01
2.6 SV對NLRP3炎癥小體相關蛋白的影響
Western blotting法檢測各組小鼠肺組織中NLRP3炎癥小體相關蛋白NLRP3、CASPASE-1、IL-18以及IL-1β的表達情況,結果如圖5所示。與CON組相比,LPS組NLRP3蛋白表達顯著性增多(P<0.01),CASPASE-1、IL-18以及IL-1β蛋白表達均顯著性增多(P<0.05);與LPS組相比,LPS+SV組中NLRP3蛋白表達明顯下調(P<0.001)、CASPASE-1蛋白表達明顯減少(P<0.05)、IL-18蛋白表達顯著性降低(P<0.01)、IL-1β蛋白的表達變化并沒有降低(P>0.05),LPS+OLT組中的IL-1β蛋白表達顯著性降低(P<0.05)。此結果說明,SV能夠部分抑制NRLP3炎癥小體的活化。
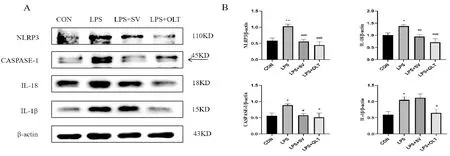
與CON組比較,**P<0.01;與LPS組比較,#P<0.05,##P<0.01,###P<0.001
2.7 SV對LPS誘導的小鼠肺組織病理學改變的影響
光學顯微鏡下觀察,HE染色結果顯示(圖6,封二):CON組肺泡結構清晰完整,無異樣;LPS組出現明顯病理學改變,包括炎性細胞浸潤、肺泡水腫、肺泡壁增厚以及肺泡紊亂;而LPS+SV組以及LPS+OLT組明顯抑制了肺組織的病理學改變。
3 討 論
急性肺損傷是指在嚴重感染、創傷、燒傷、休克等各種致病因素下誘發的急性、進行性呼吸功能不全等癥狀,其主要病理改變為肺泡-毛細血管通透性增加、炎性細胞聚集,臨床上會出現明顯的呼吸窘迫以及頑固性低氧血癥。因其發病機制極為復雜,急性肺損傷仍然是現代危重病醫學的重大難題,目前尚無有效的治療方法。
據報道[7],炎癥在ALI中起著關鍵作用,炎癥小體是炎癥相關疾病的重要治療靶點。炎癥小體是由多種蛋白質組成的復合體,其中NLRP3炎癥小體是當代研究熱點之一,可以被細菌脂多糖(LPS)、K+外流、Ca2+信號、活性氧(ROS)、線粒體功能障礙和溶酶體破裂等多種成分活化[8]。NLRP3受體蛋白含有PYD、NACHT、LRR三個結構域。當激動劑接觸受體蛋白時,NLRP3的PYD結構域與ASC的PYD結構域結合,進而ASC中的CARD結構域與效應蛋白CASPASE-1的CARD結構域結合,從而活化NLRP3炎癥小體,釋放IL-1β和IL-18,誘導免疫細胞的浸潤及其他細胞因子的產生,導致相應組織發生炎性損傷[9]。相關研究[10]表明,通過生物信息學預測,含有3 (NLRP3)的類節點受體被認為是miR-495的一個可能靶點,miR-495啟動子的甲基化可以抑制miR-495的表達,過表達miR-495可以抑制NLRP3炎性小體的激活,最終抑制LPS誘導的ALI的發展。Zhang 等研究[11]顯示,在發生急性肺損傷的小鼠體內,NLRP3炎癥小體被顯著激活并釋放了大量的IL-1β和有活性的CASPASE-1,褪黑素能夠通過抑制細胞外組蛋白而抑制NLRP3炎癥小體的激活;Cao等[12]認為,促紅細胞生成素可以通過抑制EPOR/JAK2/STAT3信號通路有效抑制NLRP3炎癥小體活化來減輕LPS誘導的急性肺損傷;枸杞多糖通過介導SIRT1抑制NLRP3炎癥小體的活化而減輕高氧誘導的急性肺損傷[13]。以上研究充分證明了抑制NLRP3炎癥小體的活化是治療急性肺損傷的關鍵。本研究結果顯示,在LPS誘導的急性肺損傷小鼠體內,NLRP3、CASPASE-1等蛋白表達明顯增多,NLRP3炎癥小體明顯被活化,釋放的IL-18以及IL-1β介導了急性肺損傷的發生發展。應用NLRP3特異性抑制劑預處理后,顯著降低了NLRP3、CASPASE-1、IL-18以及IL-1β等炎癥小體相關蛋白的表達,進一步改善了肺組織的炎癥浸潤,從而對急性肺損傷起到保護作用。
作為中性粒細胞彈性蛋白酶抑制劑,SV可競爭性抑制中性粒細胞的活化,減少急性呼吸窘迫綜合征過程中的中性粒細胞浸潤,從而對肺部起到保護作用[14]。近年來,國內外多個動物肺損傷模型均表明SV對肺損傷有保護作用,主要機制是減少肺泡中以中性粒細胞為主的炎癥細胞浸潤,抑制NE產生,并改善肺損傷的癥狀[15-16],但其與NLRP3炎癥小體的作用尚未明確。本研究結果提示,SV能夠顯著降低LPS引起的肺損傷小鼠的濕干比,明顯改變肺組織的炎癥浸潤、中性粒細胞聚集等病理學改變,抑制肺組織中p-NF-κB、NLRP3、CASPASE-1、IL-18以及NE蛋白的表達水平,對LPS誘導的急性肺損傷起到保護作用,但不能減低肺組織中IL-1β蛋白的表達水平。這些結果表明,SV作為中性粒細胞彈性蛋白酶抑制劑,能夠抑制NF-κB介導的p-NF-κB/NLRP3/CASPASE-1/IL-18通路,部分緩解急性肺損傷引起的NLRP3炎癥小體活化。
綜上所述,SV不僅能通過抑制中性粒細胞彈性蛋白酶的作用來減輕LPS誘導的急性肺損傷,還能通過部分抑制NLRP3炎癥小體活化而緩解急性肺損傷引起的炎癥作用。
猜你喜歡
興趣閱讀·興趣作文與閱讀(低年級)(2025年8期)2025-08-18 00:00:00
東方少年·布老虎畫刊(2023年8期)2023-08-01 15:45:12
科學大眾(2021年6期)2021-07-20 07:42:44
科學(2020年3期)2020-11-26 08:18:30
學苑創造·A版(2020年9期)2020-10-13 09:41:02
娃娃樂園·綜合智能(2019年3期)2019-04-03 09:17:36
中成藥(2018年2期)2018-05-09 07:19:34
小學生學習指導(低年級)(2017年10期)2017-10-10 01:00:05
湖北師范大學學報(自然科學版)(2015年2期)2016-01-10 08:41:55
云南中醫學院學報(2014年3期)2014-07-31 18:57:34
