O-GlcNAc修飾調控NLRP3炎癥小體對H9c2大鼠心肌細胞缺氧復氧損傷的影響*
2021-12-30 05:22:34張麗娜謝中杰張振剛李如君
中國病理生理雜志 2021年12期
關鍵詞:水平
張麗娜, 謝中杰, 金 慧, 張振剛, 李如君△
(1揚州大學附屬醫院心內科,江蘇揚州225001;2臺州市第一人民醫院心內科,浙江臺州318020)
以急性心肌梗死為代表的心血管疾病已成為威脅國民健康的主要病因之一[1]。雖然心肌再灌注治療已普遍開展,但臨床上仍存在恢復血供的心臟收舒功能惡化和惡性心律失常等損傷加重的現象,這與心肌缺血再灌注損傷(myocardial ischemia reperfusion injury,MIRI)密切相關[2-3]。研究證實,心肌再灌注時核苷酸結合寡聚化結構域樣受體蛋白3(nucleotide-binding oligomerization domain-like receptor protein 3,NLRP3)炎癥小體[包括含caspase 募集結構域的凋亡相關斑點樣蛋白(apoptosis-associated speck-like protein containing a caspase recruitment domain,ASC)和caspase-1]活化并引發以白細胞介素(interleukin,IL)-1β/18 產生為特征的強烈無菌性炎癥反應,是導致MIRI 的重要病理生理學基礎,因而調控NLRP3 炎癥小體可能是減輕MIRI 的新型手段[4-5]。我們的前期實驗表明,增強細胞或組織中O-連接的N-乙酰葡萄糖胺(O-linkedN-acetylglucosamine,O-GlcNAc)修飾(O-GlcNAcylation)水平,在急性應激損傷過程中具有顯著的抑制炎癥反應的作用[6]。但O-GlcNAc 修飾是否通過調控 NLRP3 炎癥小體發揮抗炎作用以及對MIRI 的影響尚不清楚。因此,本研究通過H9c2 大鼠心肌細胞缺氧再復氧(hypoxia/reoxygenation,H/R)損傷建立MIRI 的細胞模型,通過重組腺病毒感染細胞方式過表達OGlcNAc 轉移酶(O-GlcNAc transferase,OGT)以提高細胞內O-GlcNAc 修飾水平,探討其對細胞H/R 的影響及可能機制,以期為MIRI 的預防和治療提供潛在的分子干預靶標和參考資料。
材料和方法
1 材料
H9c2 大鼠心肌細胞購自中國科學院上海生命科學院細胞庫;OGT 過表達腺病毒(Ad-OGT)和空載對照腺病毒(Ad-Null)為本實驗室保存;Trizol 和RIPA 裂解液購自中國北京普利萊基因技術有限公司;O-GlcNAc(CTD110.6)抗體、OGT 抗體、GAPDH 抗體、IgG 和 Protein A/G plus Agarose 購自 Santa Cruz;IL-1β、IL-18 抗體和 HRP 標Ⅱ抗購自 Cell Signaling Technology;NLRP3 抗體、ASC 抗體和 caspase-1 抗體購自 Abcam;脂多糖(lipopolysaccharide,LPS)、TUNEL 凋亡試劑盒、DAPI 和BCA 蛋白定量試劑盒購自中國上海碧云天生物技術有限公司;增強型ECL 化學發光液、反轉錄cDNA 合成試劑盒、SYBR Green 熒光定量試劑盒、DMEM 培養液和胎牛血清購自Thermo;PCR 引物由中國上海生工生物工程有限公司合成;RT-qPCR 擴增儀(ABI 7500);Wester blot 成像系統(iBrightCL 1500)。
2 方法
2.1 H9c2大鼠心肌細胞培養 凍存的H9c2大鼠心肌細胞復蘇后用含10%胎牛血清的DMEM 培養液重懸并置于體積分數5% CO2、常氧、37 ℃培養箱中絕對靜置培養,待細胞融合度達80%左右時用0.25%胰酶消化液消化傳代,選用對數生長期的細胞行相應實驗。
2.2 病毒預處理 將細胞傳至3.5 cm 直徑培養皿培養至融合度約50%時,取感染復數為20(MOI=20)劑量的Ad-OGT 感染細胞48 h 以提高細胞內O-GlcNAc修飾水平,并設置Ad-Null感染組作為對照。
2.3 實驗分組 將感染病毒后細胞分為原生OGlcNAc修飾水平的常氧對照組(Ad-Null+Ctrl組)、H/R 組(Ad-Null+H/R 組)及高O-GlcNAc 修飾水平的常氧對照組(Ad-OGT+Ctrl 組)、H/R 組(Ad-OGT+H/R組)。通過將病毒預處理的細胞血清饑餓過夜后放置于5%CO2、94%N2、37 ℃培養箱中缺氧6 h,移置于5% CO2、常氧、37 ℃培養箱中復氧 12 h 建立 H/R模型[5]。
2.4 LPS 誘導NLRP3 炎癥小體表達 將病毒預處理的細胞血清饑餓過夜,分為原生O-GlcNAc 修飾水平的對照組(Ad-Null+Veh 組)、LPS 刺激組(Ad-Null+LPS 組)及高O-GlcNAc 修飾水平的LPS 刺激組(Ad-OGT+LPS 組)。參考已有文獻中LPS 使用濃度[7],以0.1 mg/L 終濃度的LPS 處理細胞12 h 以誘導NLRP3炎癥小體的表達,再次驗證O-GlcNAc 修飾對NLRP3炎癥小體的影響。
2.5 RT-qPCR 檢測細胞炎癥細胞因子mRNA 表達 采用Trizol 按說明提取各組細胞總RNA。紫外分光光度計測定濃度和純度后,各組取600 ng 總RNA為模板反轉錄為cDNA,行RT-qPCR。引物序列如 下 :IL-1β 的 上 游 5′-GGCGGTTCAAGGCATAACAGGCT-3′,下 游 引 物 序 列 為 5′-CAGCCCAAGTCAAGGGCTTGGA-3′;IL-18 的上游引物序列為 5′-AAGAACAAGATCATTTCCTTTGAGGA-3′,下游引物序 列 為 5′-GGAACACGTTTCTGAAAGAATATGAG-3′;18S(內參照)的上游引物序列為5′-GAAACGGCTACCACATCC-3′,下 游 引 物 序 列 為 5′-CACCAGACTTGCCCTCCA-3′。PCR 結果采用 2-ΔΔCt法進行分析。
2.6 TUNEL 染色檢測細胞凋亡損傷 采用一步法TUNEL細胞凋亡檢測試劑按說明對細胞進行凋亡染色,DAPI 復染細胞核,在熒光倒置顯微鏡下觀察,每組細胞隨機記錄4 個視野的畫面并計算凋亡陽性細胞比率。
2.7 Western blot檢測細胞蛋白表達 采用RIPA 裂解液提取細胞總蛋白經BCA 法定量后,每樣品取20 μg 總蛋白進行SDS-PAGE(10%分離膠、5%濃縮膠)電轉至PVDF 膜上,5%脫脂奶粉常溫封閉90 min,分別加入相應Ⅰ抗4 ℃孵育過夜,洗膜后用HRP 標記相應Ⅱ抗常溫孵育90 min,ECL 化學發光后使用iBrightCL 1500曝光顯影;ImageJ軟件進行定量分析,結果以目的條帶與內參條帶灰度的比值顯示。
2.8 蛋白質免疫共沉淀(Co-IP)檢測細胞內蛋白相互作用 采用Co-IP 裂解液提取細胞總蛋白經BCA法定量后,每樣品取600 μg 總蛋白分別使用NLRP3和ASC 抗體進行特異性免疫吸附,再經與Protein A/G plus Agarose 結合后離心共沉淀,將共沉淀產物清洗后經上樣緩沖液100 ℃變性后離心,取上清后行Western blot 檢測目的蛋白表達,同時設normal mouse IgG IP陰性對照。
3 統計學處理
采用SPSS 16.0軟件進行統計分析,數據結果以均數±標準誤(mean±SEM)表示,兩組間均數比較采用t檢驗,以P<0.05為差異有統計學意義。
結 果
1 H/R 對H9c2 大鼠心肌細胞內O-GlcNAc 修飾水平的影響
與 Ad-Null+Ctrl 組相比,Ad-Null+H/R 組細胞內OGT 表達及O-GlcNAc 修飾水平顯著降低(P<0.05);Ad-OGT+Ctrl 組 OGT 及O-GlcNAc 修飾水平顯著高于Ad-Null+Ctrl 組(P<0.05);且Ad-OGT+H/R 組OGT 及O-GlcNAc 修飾水平也顯著高于Ad-Null+H/R 組(P<0.05),見圖1。
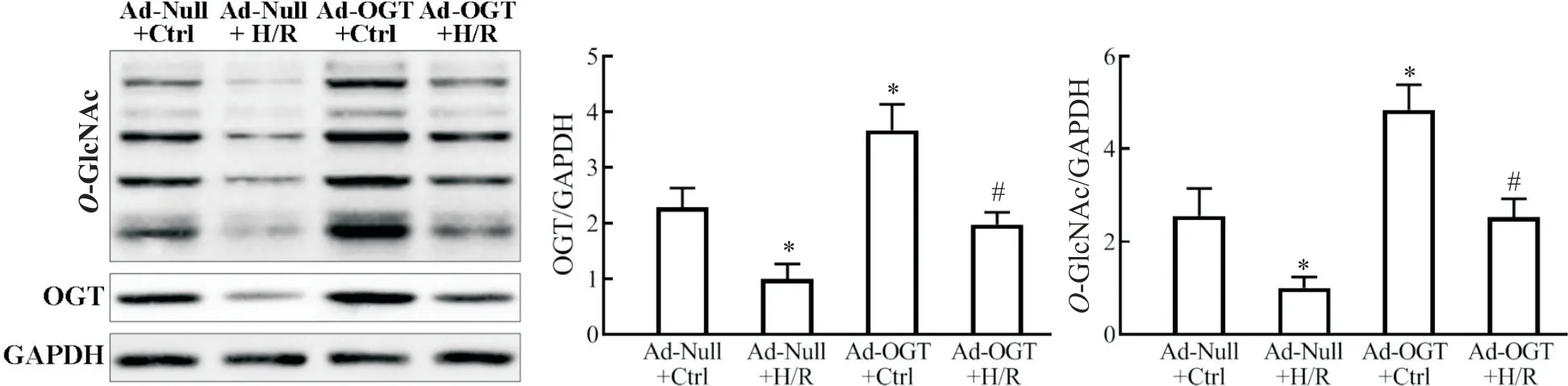
Figure 1. Over-expression of OGT increase global O-GlcNAcylation level in the cells. The levels of O-GlcNAc and OGT were detected by Western blot. Mean±SEM. n=3.*P<0.05 vs Ad-Null+Ctrl group;#P<0.05 vs Ad-Null+H/R group.圖1 過表達OGT提高細胞全局O-GlcNAc修飾水平
2 O-GlcNAc 修飾對H/R 介導細胞炎癥反應和細胞凋亡的影響
與 Ad-Null+Ctrl 組相比,Ad-Null+H/R 組炎癥細胞因子 IL-1β 和 IL-18 mRNA 大量轉錄合成(P<0.05),并出現顯著細胞凋亡現象,細胞凋亡率由(2.6±0.8)%上升至(62.8±4.2)%;Ad-OGT+H/R 組IL-1β 和 IL-18 轉錄 水 平 顯 著 低 于 Ad-Null+H/R 組(P<0.05),且細胞凋亡率由(62.8±4.2)%下降至(27.4±3.9)%,見圖2、3。
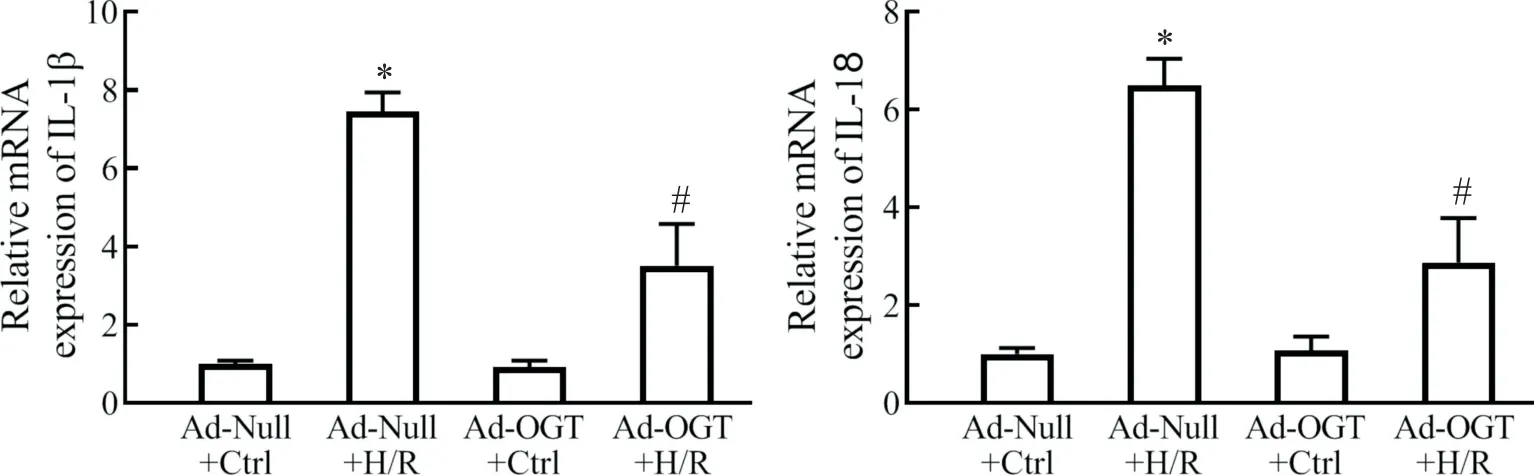
Figure 2. Increased O-GlcNAcylation level alleviated H/R-induced IL-1β and IL-18 mRNA expression. The IL-1β and IL-18 mRNA expression levels in H9c2 cells were detected by RT-qPCR. Mean±SEM. n=5.*P<0.05 vs Ad-Null+Ctrl group;#P<0.05 vs Ad-Null+H/R group.圖2 提高細胞內O-GlcNAc修飾水平可降低H/R誘導的IL1-β和IL-18 mRNA表達
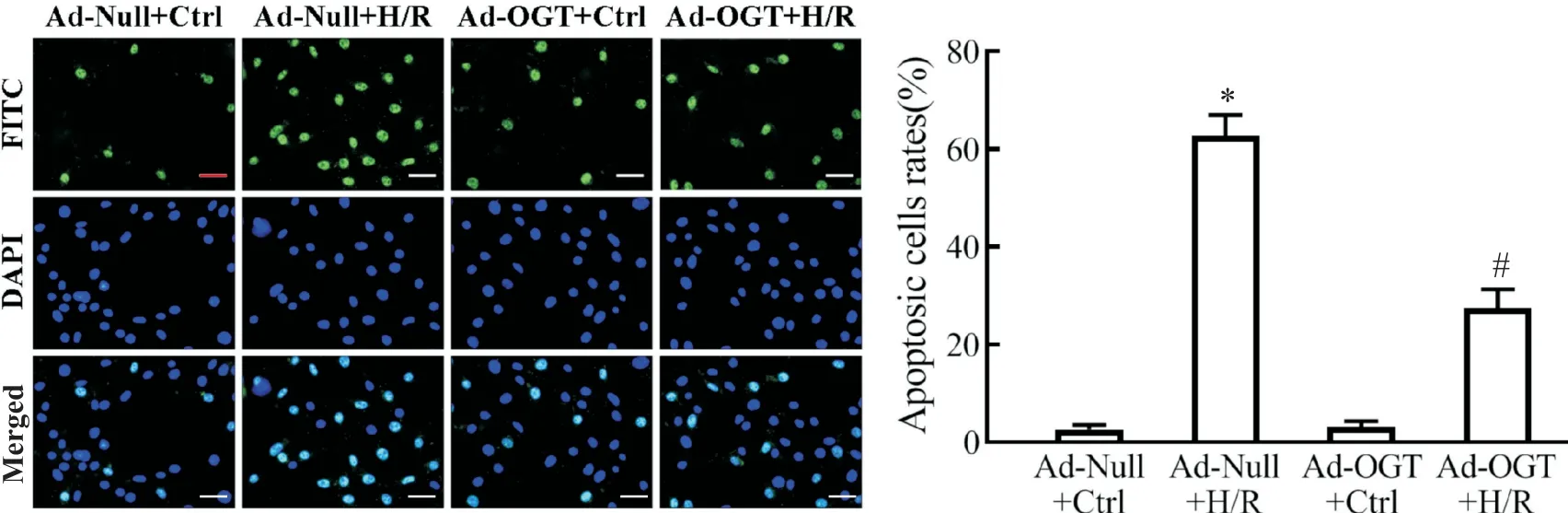
Figure 3. Increased O-GlcNAcylation level alleviated H/R-induced cell apoptosis. Apoptosis was detected by TUNEL-FITC(green)and DAPI(blue)staining. The scale bar=25 μm. Mean±SD. n=5.*P<0.05 vs Ad-Null+Ctrl group;#P<0.05 vs Ad-Null+H/R group.圖3 提高細胞內O-GlcNAc修飾水平降低H/R誘導的細胞凋亡
3 O-GlcNAc 修飾對 H/R 介導 NLRP3 炎癥小體表達的影響
與 Ad-Null+Ctrl 組相比,Ad-Null+H/R 組 NLRP3炎癥小體組成蛋白NLRP3、ASC 和caspase-1 及其效應產物IL-1β 和IL-18 表達增多(P<0.05);Ad-OGT+H/R 組 NLRP3、ASC 和 caspase-1 表 達 水 平 與 Ad-Null+H/R 組比較并統計學差異(P>0.05),但炎癥細胞因子 IL-1β 和 IL-18 表達水平較 Ad-Null+H/R 組顯著降低(P<0.05),見圖4。
4 O-GlcNAc 修飾對NLRP3 炎癥小體功能狀態的影響
與 Ad-Null+H/R 組相比,Ad-OGT+H/R 組與 NLRP3 相結合的 ASC 和 caspase-1 顯著減少,且 NLRP3免疫沉淀物O-GlcNAc 修飾水平升高(P<0.05);而兩組中ASC 免疫沉淀物O-GlcNAc 修飾水平無顯著差異(P>0.05),未檢測到O-GlcNAc 修飾的 NLRP3 信號條帶,見圖5。
5 再次驗證O-GlcNAc 修飾對NLRP3 炎癥小體表達和功能狀態的影響
與 Ad-Null+Veh 組相比,Ad-Null+LPS 組和 Ad-OGT+LPS 組均觀察到 NLRP3、ASC 和 caspase-1 的大量誘導表達(P<0.05),3 種蛋白的表達水平在后兩組間并無統計學差異(P>0.05),見圖6。而與Ad-Null+LPS 組相比,Ad-OGT+LPS 組與 NLRP3 相結合的ASC 和caspase-1減少,免疫沉淀物的O-GlcNAc 修飾水平升高(P<0.05);兩組間ASC 免疫沉淀物的OGlcNAc 修飾水平無顯著差異(P>0.05),并在NLRP3分子量118 kD附近位置無信號條帶,見圖7。
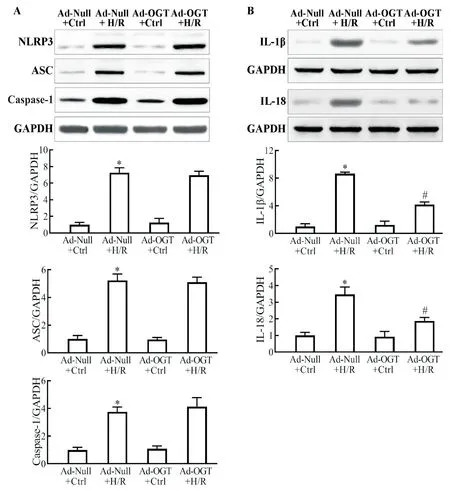
Figure 4. Increased O-GlcNAcylation level by over-expression of OGT had no effect on the expression of NLRP3,ASC and caspase-1(A)but reduced the expression of IL-1β and IL-18(B)induced by H/R injury. The levels of NLRP3,ASC,caspase-1,IL-1β and IL-18 were detected by Western blot. Mean±SEM. n=5.*P<0.05 vs Ad-Null+Ctrl group;#P<0.05 vs Ad-Null+H/R group.圖4 提高細胞內O-GlcNAc修飾水平對H/R誘導的NLRP3、ASC、caspase-1、IL-1β和IL-18表達的影響
討 論
NLRP3 炎癥小體是由 NLRP3、ASC 和 caspase-1蛋白所形成的大型蛋白復合體,在心肌缺血及再灌注過程中可被誘導表達和活化,并促使IL-1β 和IL-18 成熟和分泌[8],所引發的炎癥反應和細胞焦亡已被證實在MIRI 及實質性臟器缺血再灌注損傷發生發展過程中發揮重要作用[4,9]。但完全敲除NLRP3或阻斷NLRP3炎癥小體的表達卻觀察到了心肌梗死面積增大這一負面結果[10-11]。因而除了調節表達含量手段外,如何快速精細調控NLRP3 炎癥小體功能成為聚焦重點。
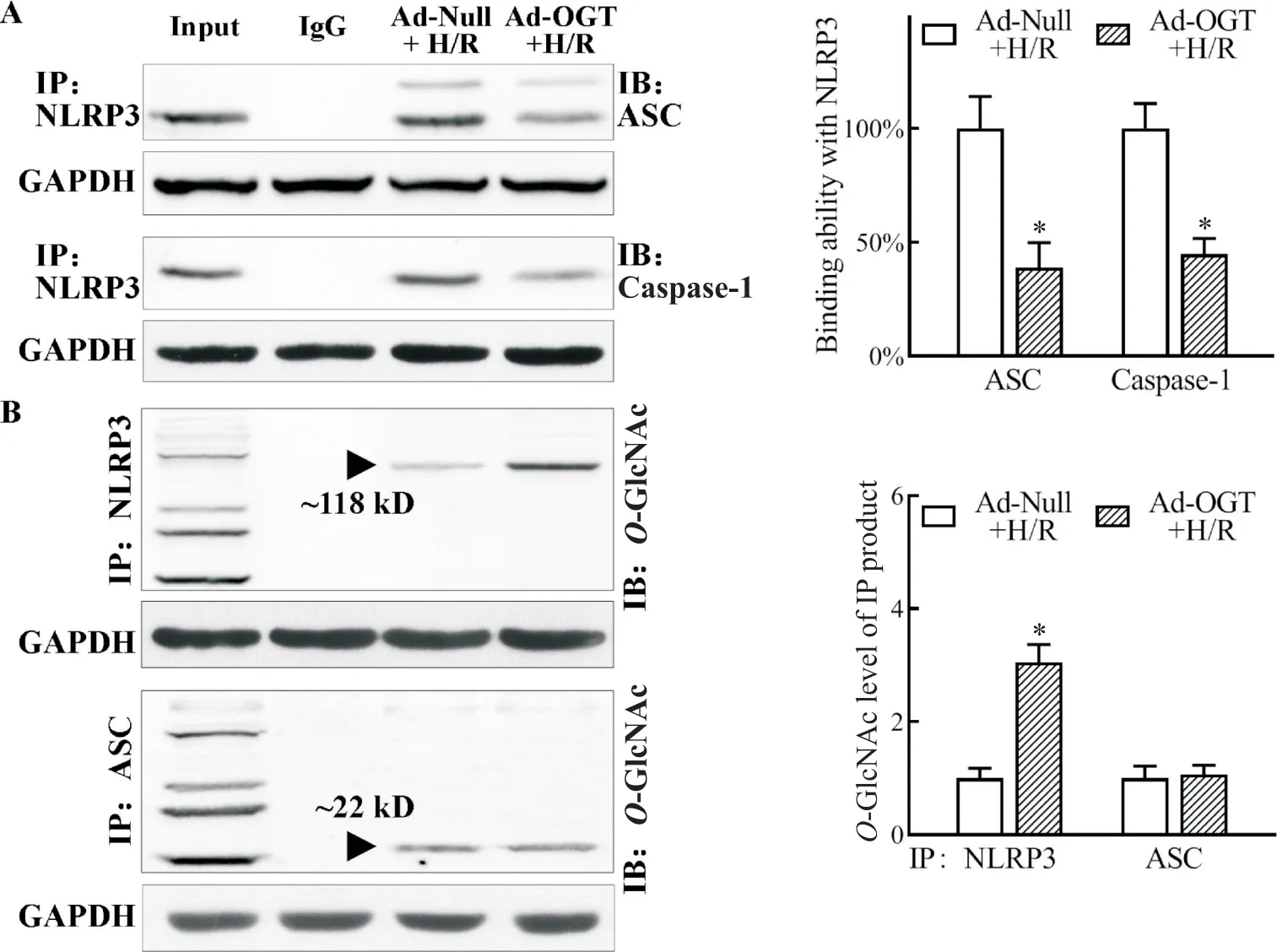
Figure 5. Increased O-GlcNAcylation level prevented the interactions of ASC and caspase-1 with NLRP3 induced by H/R injury(A)and directly increased the O-GlcNAcylation level of NLRP3 instead of ASC(B). The binding degree of ASC and caspase-1 with NLRP3 and modified status of NLRP3 and ASC were detected by Co-IP. Mean±SEM. n=3.*P<0.05 vs Ad-Null+H/R group.圖5 提高細胞內O-GlcNAc 修飾水平可直接增強NLRP3 的O-GlcNAc 修飾并阻礙H/R 誘導的ASC、caspase-1 和NLRP3 相結合
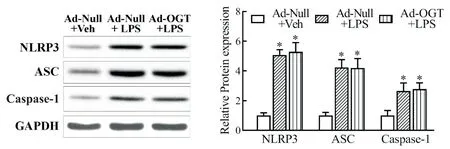
Figure 6. Increased O-GlcNAcylation level had no effect on the expression of NLRP3,ASC and caspase-1 induced by LPS. The protein levels of NLRP3,ASC and caspase-1 were detected by Western blot. Mean±SEM. n=5.*P<0.05 vs Ad-Null+Veh group.圖6 提高細胞內O-GlcNAc修飾水平對LPS誘導NLRP3、ASC和caspase-1表達的影響
本研究結果表明,提高H9c2大鼠心肌細胞內OGlcNAc 修飾水平,雖沒有抑制H/R 誘導的NLRP3 炎癥小體組成蛋白NLRP3、ASC 和caspase-1的表達,但顯著降低了NLRP3 炎癥小體活化后效應產物IL-1β和IL-18 的表達,從而緩解了H/R 誘導的炎癥反應及細胞凋亡,提示NLRP3 炎癥小體活化及其生物學功能受到負性調控。免疫共沉淀證實,提高O-GlcNAc修飾水平導致NLRP3 與ASC 和caspase-1 的相互結合受阻。由于NLRP3 只有與ASC 和caspase-1 相繼結合并形成蛋白復合體(炎癥小體)才能活化并發揮生物學功能,因此這種負性調控作用與阻礙三者相互結合密切相關。為驗證是否因NLRP3炎癥小體某種組分發生O-GlcNAc 修飾所致,我們進一步檢測了NLRP3 免疫共沉淀物(由NLRP3 抗體沉淀)的OGlcNAc修飾水平,顯示沉淀物的O-GlcNAc修飾程度隨著細胞內O-GlcNAc 修飾水平提高直接得到增強;而NLRP3 炎癥小體另一主要組分ASC 沉淀物(由ASC 抗體沉淀)的O-GlcNAc 修飾水平無顯著變化,初步表明NLRP3發生O-GlcNAc 修飾的可能性最大;且未檢測到與ASC相結合的沉淀物中存在NLRP3分子量的O-GlcNAc 信號條帶,提示NLRP3 發生OGlcNAc 修飾很可能阻礙了NLRP3 炎癥小體的組裝激活。我們再次利用經典的TLR4 激動劑LPS 誘導NLRP3 炎癥小體表達活化,同樣驗證了提高OGlcNAc 修飾水平對 NLRP3 與 ASC 和 caspase-1 相互結合的抑制作用,以及對NLRP3蛋白O-GlcNAc 修飾水平的直接提升作用。以上結果表明,除了經典的表達豐度調控方式外,通過調節翻譯后修飾(posttranslational modifications,PTMs)狀態,也是 NLRP3炎癥小體一種快速而重要的調控方式。已證實NLRP3 炎癥小體可受到多種PTMs 的調控,如多種酪氨酸激酶 Syk、Jnk 和 Btk 介導的 ASC 磷酸化能夠促進NLRP3 炎癥小體的活化[12-13];受體 TLR4 通過激活髓分化因子88(myeloid differentiation factor 88,MyD88)促進NLRP3 去泛素化是NLRP3 炎癥小體活化的重要起始環節[14];E3 泛素連接酶 FBXL2 通過促進 NLRP3 Lys689位點泛素化降解從而抑制NLRP3 炎癥小體的組裝[15],但NLRP3炎癥小體能否被O-GlcNAc 修飾尚未明確報道。
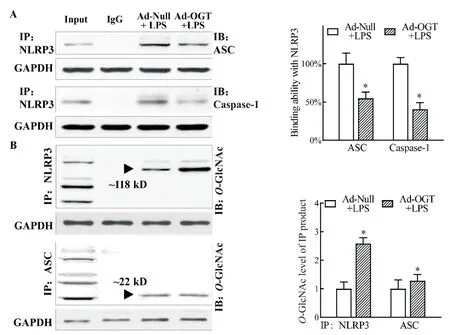
Figure 7. Increased O-GlcNAcylation level prevented the interactions of ASC and caspase-1 with NLRP3 induced by LPS(A)and directly increased the O-GlcNAcylation level of NLRP3 so that this modified status cannot be combined with ASC(B). The binding degree of ASC and caspase-1 with NLRP3 and modified status of NLRP3 and ASC were detected by Co-IP. Mean±SEM. n=3.*P<0.05 vs Ad-Null+LPS group.圖7 提高細胞內O-GlcNAc 修飾水平通過直接增強NLRP3 的O-GlcNAc 修飾并阻礙LPS 誘導的ASC、caspase-1 和NLRP3相結合
同磷酸化修飾、泛素化修飾一樣,O-GlcNAc 修飾廣泛發生于生物體內,不同的是參與O-GlcNAc 修飾調控的酶現已發現的僅有OGT 和OGA(O-乙酰氨基葡萄糖苷酶)兩種。OGT 能夠催化GlcNAc 的供體二磷酸尿嘧啶GlcNAc(uridine diphosphateN-acetylglucosamine,UDP-GlcNAc)以氧-糖苷鍵的形式共價結合到底物蛋白絲氨酸/蘇氨酸(Ser/Thr)側鏈羥基上從而增加底物O-GlcNAc修飾水平;而OGA可特異性水解氧-糖苷鍵降低底物O-GlcNAc 修飾水平[16],因而本研究采用過表達OGT 以達到提高O-GlcNAc 修飾水平的目的。
在多種應激損傷過程中均觀察到機體OGlcNAc 修飾水平的快速變化,是公認的細胞內營養及應激壓力感受器[17]。通過短期內快速提高機體OGlcNAc 修飾水平,表現出細胞保護作用,能夠增強機體對應激損傷的抗打擊能力[18-19]。我們課題組前期研究顯示,短期內提高細胞或組織的O-GlcNAc 修飾水平可通過調節NF-κB 炎癥信號通路中的多個環節來發揮抑制炎癥應答的作用。例如,直接增強NF-κB p65亞基的O-GlcNAc修飾水平能夠干擾p65的磷酸化激活[20];能夠增強抗炎因子 A20 對 NF-κB 炎癥信號通路的負反饋抑制作用[6]。國外有學者進一步證實O-GlcNAc修飾的p65亞基無法核內移并影響靶基因轉錄,減少組織中 IL-1β 和 TNF-α 的表達,顯著減少大腦缺血再灌注損傷后的腦梗死面積[21]。NLRP3 炎癥小體作為NF-κB 炎癥信號通路的重要組成環節[22],能夠直接促進炎癥細胞因子 IL-1β 和 IL-18的表達和分泌,而通過上調NLRP3的O-GlcNAc 修飾水平能夠抑制IL-1β、IL-18 mRNA 的轉錄進一步表明NLRP3 炎癥小體還可能是O-GlcNAc 修飾調控NF-κB炎癥信號通路的又一潛在靶點。
綜上所述,在本研究中我們首次獲得了NLRP3被O-GlcNAc 修飾的證據。NLRP3 發生O-GlcNAc 修飾后可阻礙H/R 誘導的NLRP3 炎癥小體的組裝活化,減少效應產物IL-1β 和IL-18的表達,從而緩解細胞的炎癥損傷。但由于免疫沉淀除NLRP3外還包含了與其相結合的其它蛋白,且尚未開發出直接檢測NLRP3 發生O-GlcNAc 修飾的方式,因此我們課題組接下來將進一步驗證NLRP3 炎癥小體活化時發生O-GlcNAc 修飾的相關蛋白及可能位點。期望為心肌缺血再灌注損傷的預防與治療提供可能的干預靶標和參考資料。
猜你喜歡
美與時代·美術學刊(2022年3期)2022-04-27 01:18:15
火花(2019年12期)2019-12-26 01:00:28
人大建設(2019年6期)2019-10-08 08:55:48
人大建設(2019年12期)2019-05-21 02:55:32
雜文月刊(2018年21期)2019-01-05 05:55:28
人大建設(2017年6期)2017-09-26 11:50:44
學苑創造·A版(2015年11期)2016-01-14 09:03:27
俄羅斯問題研究(2012年1期)2012-03-25 09:54:45
中國火炬(2010年12期)2010-07-25 13:26:22
中國火炬(2010年8期)2010-07-25 11:34:30
