硫化鋰正極材料及研究進展
2022-01-07 08:48:10蘇冠睿張春飛艾旺圣袁金良
電源技術 2021年12期
關鍵詞:復合材料
蘇冠睿, 張春飛, 艾旺圣, 袁金良
(寧波大學 海運學院,浙江寧波 315211)
鋰硫電池因具有超高的能量密度、優異的環境友好性、低廉的硫材料成本而成為下一代最具潛力的動力電池之一。鋰硫電池的應用前景廣闊,其商業化能較好地解決目前電動汽車續航里程短的問題。另外,在風能、太陽能、潮汐能等可再生能源的存儲中將發揮巨大的作用。然而,鋰硫電池也因存在很多難以克服的問題而限制其商業化應用,主要存在活性物質導電性差,多硫化物(Li2Sn)的穿梭效應及氧化還原反應速率緩慢等問題。這些問題嚴重影響鋰硫電池的比能量、庫侖效率和循環穩定性,阻礙鋰硫電池的商業化應用。鋰硫電池正極材料主要有硫單質和硫化鋰(Li2S)兩種形式,大量研究表明,Li2S 比硫具有更突出的優勢。因此,Li2S 復合正極材料的研究逐漸成為一個研究熱點,與其復合的宿主材料主要有碳基材料、聚合物、無機金屬化合物等,通過充分利用宿主材料的物理吸附和電化學催化功能,從而達到有效改善鋰硫電池性能的目的。
1 鋰硫電池正極材料及存在的主要問題
1.1 鋰硫電池正極材料
鋰硫電池屬于鋰金屬電池中的一種,其中正極材料可細分為硫基和Li2S 基兩類。電池涉及的反應機理相同,僅充放電順序不同。以硫為正極材料情況下,放電時負極的鋰失去電子變為鋰離子,正極的硫與鋰離子化合生成Li2Sn,并最終生成Li2S。在充電過程中,鋰硫電池的正負極反應逆向進行,具體反應如(1)所示。若正極為Li2S,則反應過程如(2)所示。
(1) 正極為單質S
電極反應過程[1]:
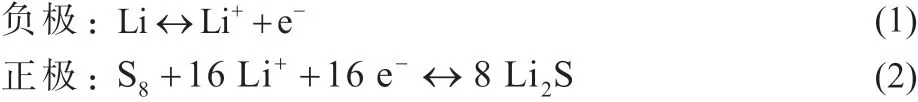
(2) 正極為Li2S
電極反應過程:

研究表明,具體的放電過程更為復雜。硫在反應中被逐步鋰化(S8→Li2S8→Li2S6→Li2S4),形成一系列Li2Sn,可溶的長鏈Li2Sn再逐步形成短鏈的不可溶硫化物(Li2S4→Li2S2→Li2S),進而以固體形式重新沉淀到電極上。由此可見,隨著反應的進行,電極反應經歷了固-液-固的轉變,是一個多相反應體系。因此,與傳統的鋰離子電池有很大不同之處。
1.2 鋰硫電池硫正極存在的主要問題
(1)導電性差:正極活性物質單質硫導電性較差,其中硫單質的電導率只有5×10-30S/cm[2]。
(2)穿梭效應:放電中間產物Li2Sn易溶解在有機電解液中,導致活性物質硫的減少。在正極溶解的長鏈Li2Sn穿過隔膜到負極與金屬鋰反應,被還原成短鏈的Li2Sn;而充電時,負極的Li2Sn又會穿過隔膜回到正極,被氧化成長鏈的Li2Sn。這個過程就是鋰硫電池的“穿梭效應”。“穿梭效應”可導致鋰硫電池中活性物質損失,金屬鋰負極被腐蝕,造成庫侖效率低和循環壽命短等問題[3]。由于存在“穿梭效應”,Li2Sn最終被還原成Li2S,并沉積在負極鋰的表面上形成鈍化層。這不僅導致活性物質的損失還增加了電池的阻抗,阻礙了負極鋰與電解液的良好接觸,從而抑制負極與電解液之間的鋰離子交換,減緩了電極的反應動力學[4]。
(3)體積膨脹:因為硫的密度為1.96 g/cm3,Li2S 的密度為1.66 g/cm3,兩者存在較大的密度差,導致單質硫在鋰化過程中產生約80% 的體積膨脹[5]。體積增大會使部分活性物質與集流體失去良好接觸,破壞電極結構,切斷電子傳輸,造成電池容量下降。
(4)電極動力學過程緩慢:硫存在電子絕緣性等多因素引起的氧化還原反應速率緩慢的問題,特別是液態Li2S4向固態Li2S 的轉化遲滯以及Li2S 較高的活化能壘,嚴重制約著電池的倍率性能[6]。
1.3 硫化鋰正極材料的主要問題及優勢
Li2S 是鋰硫電池硫正極放電的終產物,可以看作是硫的預鋰化。因此,二者存在共性問題。硫存在電子絕緣性,Li2S也是電子絕緣材料,導電率很低(電導率約10-13S/cm)[7],在其氧化過程中,也同樣存在Li2Sn的“穿梭效應”和氧化還原反應動力學緩慢等問題。區別之處是,Li2S 對水較敏感,在空氣中易吸收水蒸氣發生水解,放出劇毒硫化氫氣體。因此,Li2S 的制備條件也相對苛刻,主要采用球磨法[8]、溶劑法[9]、直接碳復合法[10]等。另外,Li2S 作為正極材料,其初始氧化時還需要克服一個很大的活化能壘(過電壓)[11]。不過,Li2S 也有很多優勢,其理論比容量高達1 166 mAh/g[12],還能避開硫的首次鋰化過程導致的體積膨脹問題。此外,它能和非鋰金屬陽極(碳、硅等)配對組成鋰離子電池,避開了一直懸而未決的金屬鋰枝晶問題。因此,Li2S 是非常有潛力的正極材料。
2 Li2S正極材料的研究進展
2.1 碳基/Li2S 復合材料
2.1.1 單質碳/Li2S 復合材料
單質碳材料的種類很多,主要有熱解碳、石墨、碳納米管、石墨烯等。碳材料具有良好的導電性,能有效地提高電極材料的導電率,從而提高電池的比容量。
熱解碳或低結晶度碳材料比較容易制得,通過簡單的高溫熱解即可形成碳/Li2S 復合材料,最具有代表性的方法是化學氣相沉積法(CVD)。Yang 等[13]通過CVD 技術在Li2S 顆粒上成功沉積均勻的導電碳層,獲得了核-殼結構的納米復合材料(nano-Li2S@C)。所制備的納米Li2S 顆粒的平均尺寸約為100 nm,碳殼厚度約為20 nm。Li2S@C 顆粒可以縮短鋰離子的擴散距離,且外殼碳層可以加速電子傳輸,并能夠有效限制可溶性多硫化物的遷移。在0.2C時初始放電比容量達到1 083.5 mAh/g,200 次循環后比容量為766.3 mAh/g,循環衰減率為0.15%。另外,高溫熱解含碳聚合物前驅體也是較常用的方法。Liang 等[14]采用聚苯乙烯為前驅體,通過高溫熱解合成出具有高Li2S 含量的多孔碳涂層復合材料Li2S@C。該復合材料在0.1 A/g 的電流密度下循環200 次,單次容量衰減率為0.18%,表現出優異的循環穩定性。
碳納米管和石墨烯具有出色的物理化學性質,因而也常被用作鋰硫電池正極的負載材料。Chen 等[15]通過噴霧干燥法和熱處理制得Li2S 與碳納米管(CNT)的復合材料(Li2S@CCNT)[見圖1(a)]。3D CNTs 骨架網絡可抑制Li2Sn的擴散,同時提供良好的電子傳輸路徑,使得Li2S@C-CNT 復合材料具有良好的倍率性能和循環穩定性,如圖1(b)、(c)所示。Li 等[16]采用熱解法將石墨烯納米片聚集體(GNA)和硫酸鋰(Li2SO4)的混合物制備成Li2S-GNA 復合材料。實驗結果表明,Li2SGNA 樣品的初始放電比容量為693 mAh/g,第40 次放電比容量為508 mAh/g,庫侖效率約為95%。 賈順新等[17]制備的Li2S/石墨烯復合材料具有豐富的微孔蜂窩狀結構,可顯著提高復合電極材料的電導率并能通過物理截留作用限制Li2Sn的擴散,從而獲得較高的庫侖效率和良好的循環性能。表1為不同碳基/Li2S 復合材料的循環性能。
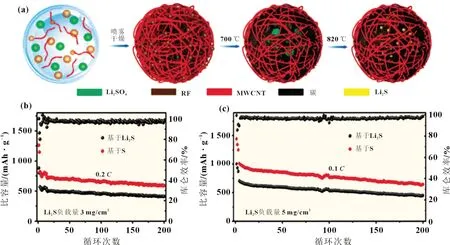
圖1 (a)Li2S@C-CNT的合成過程示意圖;(b) 電流密度為0.2 C時,負載量為3 mg/cm2的Li2S@C-CNT陰極的長期循環穩定性;(c) 在0.1 C電流密度下,負載量為5 mg/cm2的Li2S@C-CNT陰極的循環性能[15]
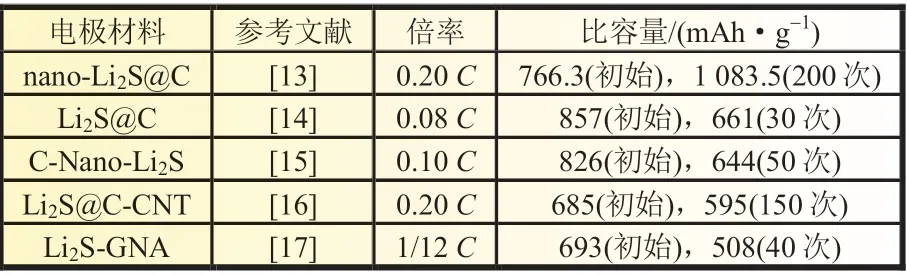
表1 不同碳基/Li2S 復合材料的循環性能
2.1.2 雜原子摻雜的碳基/Li2S 復合材料
采用雜原子摻雜的方法,可增強非極性碳材料與極性Li2Sn之間的相互作用力并顯著提升碳基材料的導電性,因此是改善Li2S 電極性能的有效手段。目前,碳材料的雜原子摻雜主要有氮原子摻雜、磷原子摻雜、硼原子摻雜、過渡金屬原子摻雜等形式。其中,氮摻雜的研究已獲得較大進展[18]。Hu等[19]在二甲基甲酰胺(DMF)溶液中蒸發聚丙烯腈(PAN)和Li2S 的混合物,成功合成了Li2S@NRC 復合材料。在0.5C的高倍率下,電池的初始比容量為958 mAh/g,超過1 000 個循環后,每個循環的容量衰減率僅為0.041%。研究發現,磷/氮共摻雜比單一氮原子摻雜效果更佳,Zhang 等[20]合成了一種來自植酸摻雜聚苯胺的氮磷共摻雜碳(N,P-C)骨架,制備無粘結劑的Li2S 陰極。摻有N 原子和P 原子的3D 多孔碳結構可以實現更好的電子傳輸和Li+遷移。P 原子摻雜的碳骨架增強了與硫的相互作用,從而有助于抑制多硫化物的溶解與擴散,提高了庫侖效率。此外,P 原子摻雜碳材料改善了反應動力學,通過催化電極反應降低了電極極化,并在提升離子電導率方面起著至關重要的作用。在0.1C的100 個循環中,電池庫侖效率為99.4%,放電比容量為700 mAh/g。硼摻雜的研究相對較少,Zhou 等[21]用液體滲透-蒸發涂覆的方法分別制備出了氮、硼摻雜的石墨烯氣凝膠負載Li2S 的復合電極材料,制備方法如圖2(a)所示。雜原子N 或B 摻雜有利于電荷快速轉移,并能改善非極性石墨烯骨架和極性Li2Sn之間的親和力,使石墨烯電極具有穩定的循環性能。從圖2(b)可知,在0.5C倍率下,相比未摻雜的還原石墨烯,二者的比容量均有較大幅度提升,放電比容量分別為657 和597 mAh/g,300 次循環后平均容量衰減率分別為0.129% 和0.134%。由此可見,N 原子摻雜要優于B 原子參雜。表2 為不同元素摻雜的碳基/Li2S 復合材料的循環性能。
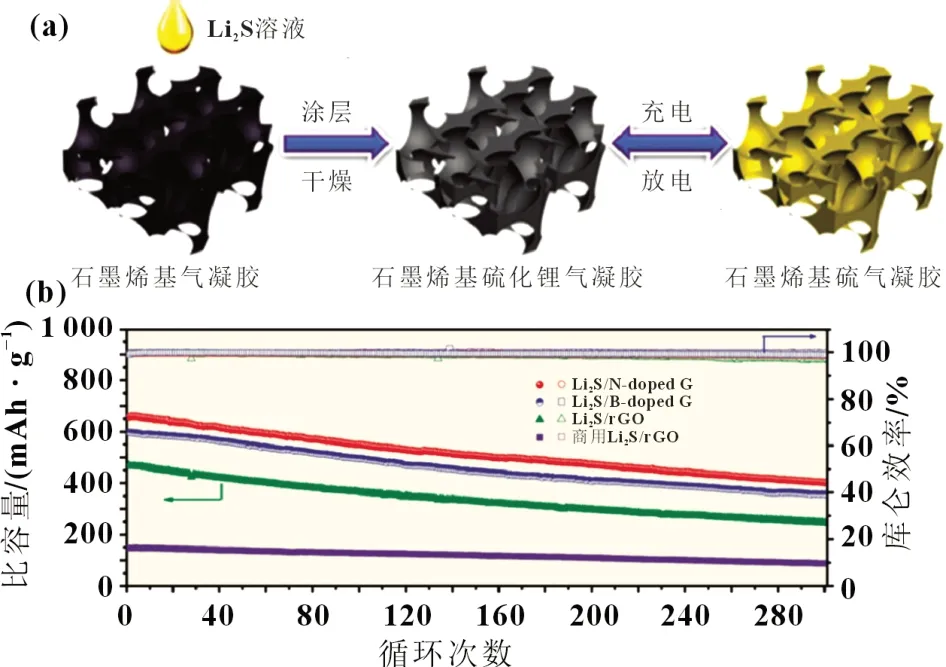
圖2 (a) Li2S的涂覆過程以及石墨烯基電極的原位充放電示意圖;(b) 在0.5 C倍率下,商用Li2S-rGO、Li2S/rGO、Li2S/B-doped G 和Li2S/N-doped G等電極的300次循環充放電性能和庫侖效率[21]

表2 不同元素摻雜的碳基/Li2S 復合材料的循環性能
2.2 聚合物基/Li2S 復合材料
聚合物用于鋰硫電池的研究相對較少。Cui 等[22]通過在Li2S 顆粒上進行吡咯的原位聚合,合成了Li2S-聚吡咯(PPy)復合材料。PPy 中的N 原子帶有孤對電子,它與Li2S 具有較強的Li-N 相互作用,可使PPy 牢固地覆蓋在Li2S 的表面,從而抑制Li2Sn在電解質中的溶解。另外,導電的PPy 也有助于電子傳導。另一類是非導電聚合物,目前很少用于鋰硫電池系統中。 Hao 等[23]選用一種具有離子選擇性的磺化聚醚醚酮(SPEEK)膜,與獨立式SWCNT/rGO 結構構成界面層[如圖3(a)]。因為SPEEK 膜帶有負電的離子納米通道,可以成為阻攔Li2Sn擴散的屏障和促進Li+離子快速傳輸的通道。從圖3(b)和(c)可知,相對未添加聚合物的參比組而言,SPEEK 聚合物明顯改善了電池的循環性能。
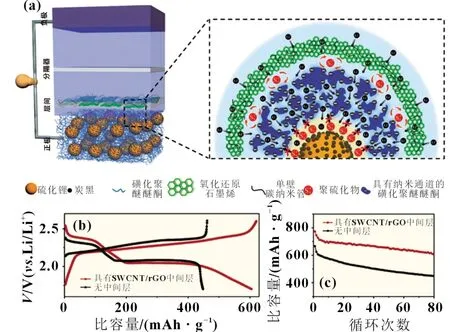
圖3 (a)含有SPEEK 的陰極與SWCNT/rGO中間層構成的界面示意圖;(b)有/無SWCNT/rGO 中間層條件下,Li2S陰極的電池電壓-比容量曲線對比圖;(c)有/無SWCNT/rGO 中間層條件下,電池的循環性能對比圖[23]
2.3 無機金屬化合物基/Li2S 復合材料
無機金屬化合物也是一類極有潛力的Li2S 宿主材料,該類化合物的極性表面與極性的Li2Sn之間有較強的相互作用(路易斯酸-堿相互作用),具有表面吸附和催化的雙重效果,因而引起了研究人員的高度重視。研究表明,利用金屬與Li2Sn之間的路易斯酸堿相互作用能有效地抑制Li2Sn的“穿梭效應”[24]。目前研究較多的主要有金屬碳(氮)化物(MXene)、金屬氧化物、金屬硫化物、金屬磷化物等。
2.3.1 MXene 基/Li2S 復合材料
MXene 因具有類似石墨烯的典型二維(2D)結構而受到廣泛的關注[25]。作為MXene 的代表化合物,過渡金屬碳化物和氮化物具有高導電性、獨特的三明治層狀結構等,已被廣泛應用于電極材料。
王等[26]合成了Li2S@MXene/G 陰極材料。多層片狀的Ti3C2材料比表面積大、表面活性位點多,不但可以增加活性物質Li2S 的負載量,還能充分起到吸附催化的效果。結果表明,Li2S@MXene/G 陰極中Li2S 的含量高達9 mg/cm2,比容量為5.04 mAh/cm2,表現出良好的循環穩定性。Liang 等[27]通過高能球磨法制備了多層的Ti3C2/Li2S 復合材料(ML-Ti3C2/Li2S),在0.2C條件下,100 次循環充放電后,能保持450 mAh/g 的電池比容量(初始電池比容量約為700 mAh/g)。 Pourali 等[28]將Ti3C2Tx分散在Li2S-EtOH 溶液中,制備了Li2S/Ti3C2Tx復合材料。電極初始比容量為708 mAh/g,100 次循環后容量保持率為74.5%(528 mAh/g)。
2.3.2 金屬氧化物/Li2S 復合材料
金屬氧化物可作為鋰硫電池中硫的宿主材料,該類研究主要通過構建不同的形貌結構(包括一維的納米線、納米棒,二維的納米片,以及三維的納米球等),從而合成出具有較大比表面積和分層結構的金屬化合物材料。近年來,金屬氧化物負載Li2S 的研究逐漸成為一個熱點。Chen 等[29]通過熱處理和原子層沉積技術制備出氧化石墨烯海綿負載氧化鋁和Li2S (Al2O3-Li2S-GS)的分層結構材料。Li2S 表面沉積的納米Al2O3不僅對Li2Sn的擴散有物理限制作用,還可以通過鋰-氧鍵合作用抑制長鏈Li2Sn的溶解,從而確保較高的比容量。Al2O3-Li2S-GS 具有736 mAh/g的初始比容量,300次循環后容量保持率為88%(0.5C),1 000 次循環充放電后容量衰減率為0.028%(2C)。Yuan 等[30]證明Li2S 與TiO2(110)晶面之間的鍵能強度(≥2.18 eV)明顯大于其與石墨烯之間的鍵能強度(<1.0 eV),所以金屬氧化物TiO2可更好地與Li2S 結合,提高Li2S 的利用率。Wu 等[31]通過蒸發含有鈦基的Li2S-乙醇溶液,然后進行熱處理,制備Li2S@LiTiO2復合材料,如圖4(a)所示。因為層狀的LiTiO2殼不僅具有離子和電子傳導性,而且能與Li2S形成非常強的化學鍵。最重要的是,LiTiO2可以將長鏈Li2Sn快速轉化為短鏈Li2Sn,表現出較高的倍率性能和循環穩定性[圖4(b)]。
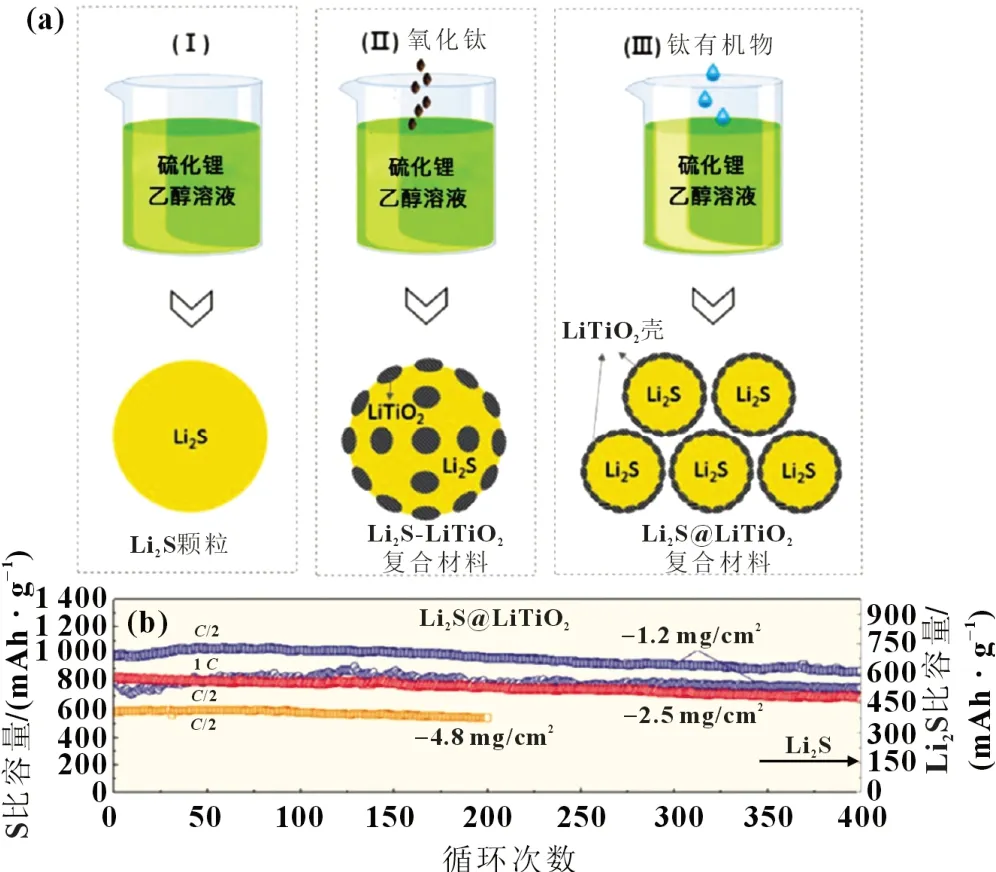
圖4 (a) Li2S、Li2S-LiTiO2和Li2S@LiTiO2復合納米顆粒的工藝流程示意圖;(b) Li2S@LiTiO2陰極在C/2和1 C下的長期循環穩定性[31]
2.3.3 金屬硫化物/Li2S 復合材料
金屬硫化物廣泛存在于自然界中,有很強的親硫性,可用作鋰硫電池硫正極的宿主材料。Seh 等[32]用二維層狀過渡金屬TiS2作為鋰硫電池陰極的宿主材料,合成了納米Li2S@TiS2核-殼高導電性復合材料[圖5(a)]。TiS2中存在極性的Ti-S 鍵,Ti 原子可以通過Ti-S 鍵合作用與Li2S 以及Li2Sn產生化學吸附作用,因此Li2S 與TiS2的結合能遠高于其與碳基石墨烯之間的結合能。如圖5(b)和(c),Li2S@TiS2材料表現出良好的倍率性能。Chung 等[33]設計了一種Li2S-二硫化鈦-電解質(Li2STiS2-E)復合陰極。這種復合結構同時提高了Li2S 的活化效率、電荷存儲容量和循環壽命。TiS2是一種可吸附Li2Sn的電化學活性材料,能有效阻止Li2Sn溶解在電解液中。另外,TiS2的高電導率可增強電池正極Li2S 的氧化還原反應速率,將Li2S-TiS2-E 陰極的活化能勢壘從3.5 V 降到3.0 V,高效活化了Li2S,促進了電極反應。因此,Li2S-TiS2-E 復合材料具有較高的質量比容量(704 mAh/g)和面積比容量(4.23 mAh/cm2)。
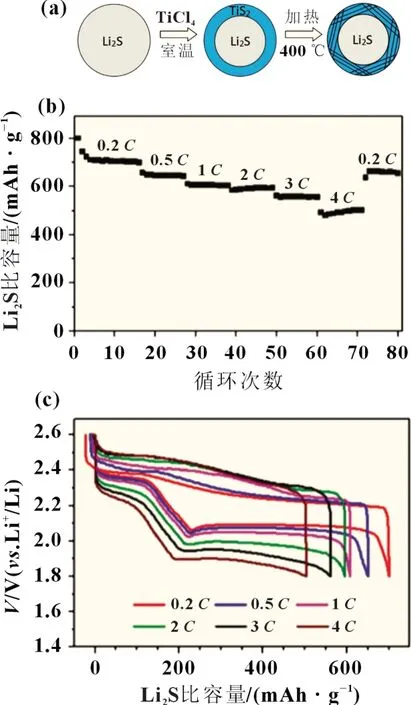
圖5 (a) Li2S@TiS2核殼納米結構合成過程的示意圖;(b)0.2 C到4 C多倍率循環充放電測試,Li2S@TiS2陰極的比容量;(c)充放電壓分布圖[32]
2.3.4 金屬氮化物/Li2S 復合材料
相對金屬氧化物和金屬硫化物而言,金屬氮化物具有更高的電子導電性,因此是十分有應用潛力的宿主材料之一。Zhou 等[5]采用石墨烯包覆具有TiN-TiO2異質結構的納米復合物作為Li2S 的宿主材料。碳的非極性表面對極性多硫化鋰(LiPS)的親和性較差,但石墨可通過物理阻隔作用抑制LiPS的擴散。TiO2和TiN 對LiPS 均有很強的吸附性,此外,TiN 還具有電催化功能,可促進LiPS 向Li2S 轉化,在電極界面上完成LiPS 的捕捉-擴散-轉化等過程,很好地抑制了“穿梭效應”,實現活性物質的高效利用和反應的快速進行。在0.3C倍率下,7TiN:3TiO2-G 涂層可提供927 mAh/g 的比容量,每個周期的容量衰減僅為0.027%。但是,由于氮化物的合成條件比較嚴苛、成本高、結構難以控制,其應用受到了較大的限制。
3 結論與展望
綜上所述,碳基材料、聚合物、金屬化合物等作為負載Li2S 的宿主材料均能不同程度地改善電池的性能。雜原子摻雜的碳基材料和無機金屬化合物是性能表現較為突出的兩類宿主材料,但二者也各有優缺點。其中,碳基材料制備工藝相對簡單,而且兼有廉價和環保的特點,因此應用潛力巨大。但是,摻雜的碳材料也存在極性較弱,振實密度低,表面活性位點數量無法有效控制的弊端。金屬化合物表面的極性活性位點與Li2Sn的相互作用,可以有效改善Li2S 正極的性能。另外,通過采用多種納米合成技術,構建分層的、高比表面積的金屬化合物材料,可以進一步提高Li2S 的負載率。相對多孔碳宿主材料而言,納米金屬化合物對Li2Sn的吸附能力更強且有較高的振實密度來維持電池結構的穩定性。但是,大多數金屬化合物導電性較差,密度大,脆性高,特別是Li2S 的電極負載率遠遠低于實際應用需求,其在鋰硫電池中的應用受到了很大的限制。
就未來的發展趨勢而言,Li2S 復合正極材料仍將是鋰硫電池研究的熱點方向之一。如何有效抑制多硫化物在電解液中的溶解和提升電極反應動力學仍是要攻克的重點問題。具體來講,宿主材料的選擇要考慮如下特點:高比表面積,以實現硫的高負載量;高電導率,以增強電極反應動力學;材料結構有利于鋰離子傳輸和硫的體積膨脹;材料表面對Li2Sn的氧化還原反應有高效催化作用等。在充分考慮以上特性的同時,盡可能提高Li2S 在復合材料中的含量,從而獲得具有長循環壽命和高能量密度的Li2S 基復合材料。如果鋰金屬問題也能得到很好解決,鋰硫電池必將在未來的新能源領域占據重要的一席之地。
猜你喜歡
建材發展導向(2022年2期)2022-03-08 01:44:04
建材發展導向(2021年14期)2021-08-23 00:56:16
中國材料進展(2019年10期)2019-12-07 05:32:14
纖維復合材料(2018年3期)2018-04-25 07:22:58
電子測試(2017年11期)2017-12-15 08:57:13
山東工業技術(2016年15期)2016-12-01 05:31:34
中國塑料(2015年6期)2015-11-13 03:02:54
中國塑料(2015年11期)2015-10-14 01:14:14
中國塑料(2015年8期)2015-10-14 01:10:41
應用化工(2014年10期)2014-08-16 13:11:29
