節段性皮膚僵硬綜合征一例
2022-01-18 08:39:06陳聲利盧憲梅陳學超
中國麻風皮膚病雜志 2022年3期
關鍵詞:特征
陳聲利 盧憲梅 劉 紅 陳學超
山東第一醫科大學附屬皮膚病醫院(山東省皮膚病醫院),山東省皮膚病性病防治研究所,濟南,250022
臨床資料患者,男,5歲。左股部皮膚硬化4年余。患者4個月時左股部皮膚發硬,逐漸擴展,曾于當地醫院就診,考慮硬皮病、嗜酸性筋膜炎、結締組織痣,未治療。患兒父母非近親結婚,足月剖腹產出生,出生體重3.5 kg。家族中無類似患者,否認遺傳病史。體格檢查:患兒一般情況良好,營養、發育及智力均正常。心肺腹部查體未見異常。左膝關節活動不受限。皮膚科檢查:左股部自腹股溝至膝上方皮膚彌漫性硬化,與下方組織粘連,不能捏起,與周圍組織分界不清,左股比右側略細,皮損表面毛發增多(圖1)。實驗室檢查:血尿常規、肝腎功能未見異常。皮損組織病理示:表皮輕度乳頭瘤狀增生,真皮膠原纖維增粗,排列紊亂,膠原纖維間可見脂肪細胞,未見明顯炎癥細胞浸潤(圖2)。阿新藍染色:真皮內黏蛋白增多(圖3)。基因檢測未發現既往報道致病基因FBN1的突變。診斷:節段性皮膚僵硬綜合征。建議患者積極進行功能鍛煉,目前仍在隨訪中。
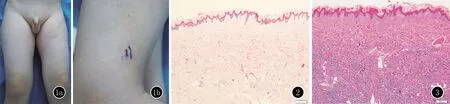
圖1 1a: 左股部皮膚硬化,比右腿略細;1b:毛發增多 圖2 表皮輕度乳頭瘤狀增生,真皮中深部膠原纖維增粗,排列紊亂,膠原纖維間見脂肪細胞(HE,×200) 圖3 真皮內黏蛋白沉積(阿新藍染色,×200)
討論皮膚僵硬綜合征(stiff skin syndrome,SSS),又稱為先天性筋膜發育不良。目前國內外報道較少,檢索PUBMED和知網,共查到約60例報道,臨床易誤診。1971年,Esterly和Mckusick[1]首次描述皮膚僵硬綜合征。國內由耿松梅于2002年首次報道[2]。SSS的病因和發病機制尚不清楚,目前認為由于fibrillin-1(FBN1)編碼的基因突變,導致轉化生長因子(TGF-β)信號活化,從而使成纖維細胞異常增殖[3]。目前所報道的存在FBN1基因突變的家系均符合常染色體顯性遺傳特征,且受累患者均屬于泛發型SSS,節段性SSS未能找到其致病突變[4]。
SSS發病年齡較早,多發于嬰兒期或兒童期。好發于臀部和股部。臨床表現為皮膚硬化,伴有輕-中度毛發增多,進展緩慢,嚴重時關節活動受限。Myers等[5]對已報道的52例SSS進行了分析,認為SSS可分為節段性SSS和彌漫性SSS兩種。節段性SSS以單側皮損為主,平均發病年齡4.1歲,男女發病率為3.5∶1;彌漫性 SSS 累及雙側,平均發病年齡1.6歲,男女發病率相同,男性略多,97%的患者關節活動受限,而節段性SSS中有44%的患者關節活動受限。平均隨訪時間11年,報道的18例節段性SSS都沒有進展為雙側受累。節段性SSS和彌漫性SSS的組織病理表現相同。SSS的病理學特征為:膠原束增厚,水平排列,缺乏炎癥,膠原間黏蛋白增多。Myers等[5]認為在膠原間出現脂肪細胞是診斷該病的一個主要特征。
SSS應與硬皮病、硬腫病、嗜酸性筋膜炎、結締組織痣鑒別。(1)硬皮病的皮損為象牙色或白色的致密硬化,無毛發增多的現象。組織病理學特征為真皮網狀層膠原纖維束致密、均質化,在早期階段,有密集的淋巴細胞、漿細胞浸潤。晚期階段,皮膚附屬器萎縮消失。(2)硬腫病多見于成人,好發于頸項和背部。組織病理學特征是真皮網狀層顯著增厚,膠原間黏蛋白沉積。(3)嗜酸性筋膜炎起病迅速,臨床特征是疼痛、觸痛性硬結,表現為凹陷性水腫、橘皮樣外觀。病理表現為淺筋膜明顯增厚、纖維化,真皮網狀層深部可出現慢性炎細胞浸潤。實驗室檢查外周血嗜酸粒細胞增多。(4)結締組織痣表現為堅實的單個或多發性呈膚色的丘疹、結節或斑塊,病理表現為增加的膠原束無規則排列。
本例患兒嬰兒期發病,發生于一側股部,臨床表現為皮膚硬化,毛發增多。病理表現為真皮網狀層膠原纖維增粗,無炎細胞浸潤,黏蛋白增多,膠原纖維間可見大小不一的脂肪團塊。基因檢測未發現FBN1的突變。綜合臨床和病理表現可排除其他皮膚硬化性疾病,符合節段性SSS的診斷。尤其是病理特征中膠原纖維間可見大小不一的脂肪團塊,與Myers等[5]對該病的病理特征的觀點一致。
皮膚僵硬綜合征病情進展緩慢,尚無有效治療方法,故以對癥治療為主。應盡早進行康復理療,以防止局部功能障礙。
猜你喜歡
數學小靈通·3-4年級(2024年2期)2024-05-15 02:02:28
中學生數理化(高中版.高考數學)(2022年3期)2022-04-26 14:04:16
數學年刊A輯(中文版)(2020年1期)2020-05-19 00:30:36
空間科學學報(2020年2期)2020-04-01 03:50:40
瘋狂英語·新策略(2019年10期)2019-12-13 08:43:28
中等數學(2019年8期)2019-11-25 01:38:14
當代陜西(2019年10期)2019-06-03 10:12:04
新聞傳播(2018年11期)2018-08-29 08:15:24
數學小靈通·3-4年級(2017年9期)2017-10-13 08:10:54
廣西科技大學學報(2016年1期)2016-06-22 13:10:38
