
基于第一性原理對尖晶石結構LiMn2O4正極材料和相關化合物進行理論研究
2022-01-22 06:05:54伍澎貴姜興濤寧玉娟黃東雪梁興華
裝備制造技術 2021年10期
關鍵詞:結構
伍澎貴,姜興濤,張 宇,寧玉娟,黃東雪,梁興華
(廣西科技大學 機械與汽車工程學院,廣西 柳州 545006)
0 引言
由于石油、天然氣等可再生能源的緊缺和環境污染不斷加劇問題的出現,研究學者們逐漸把目光投向于可再生新能源材料的研究和開發,從而帶動了新能源電車以及動力電池的發展[1]。鋰電池作為新的能源供給方式,為新能源發展的重點[2]。目前,各研究團隊對于正極材料的研究相對深入。而研究學者們現也正在尋求一種具有原材料豐富且價格便宜、對污染環境友好污染小、高能量比、高工作電壓、電化學性能好、安全性高等優點的電池正極材料。因此,研究學者們找到了一種滿足上述條件的尖晶石LiMn2O4材料,但由于其仍然存在諸多缺點,如:在充放電過程中比容量快速衰減、不耐高溫、容易出現Jahn - Teller畸變效應,在很大程度上阻礙了它的大規模商業化發展。為此,研究學者們需要找尋一種有效且合理的改良途徑去優化尖晶石LiMn2O4材料的缺點,目前主流有效的改良途徑是摻雜[3]。通過在材料中摻入適當的過渡金屬X原子代替某些Mn原子,可以有效地改善材料的電化學性能[4]。通過進行元素摻雜,提高了容量比、能量比使其穩定性能更好,元素的摻雜導致電位能表面的變化,可能會改變Li+遷移的屏障,還有可能改變Li+遷移的路徑[5],因此對LMO材料進行深入研究Li+遷移路徑顯得非常有必要。
近年來,第一性原理研究在Li+電池領域占有越來越重要的地位,通過第一性原理計算可以解釋材料的內部結構、材料摻雜前后屬性變化等信息[6]。申海燕等利用第一性原理計算了尖晶石型鋰錳氧化物的結構特征,結果表明:缺陷型LiMn2O4中理論容量隨著Li含量的增加而減小;王延慶等利用第一性原理計算了Ni -3d軌道對LiMn2O4的誘導作用,結果表明:Mn位摻雜Ni后Mn - O的鍵強增加,提高了LMO的電子導電率以及其結構的穩定性,進一步加快了Li+的擴散速度[7]。
高農等使用溫差法生成LAMO材料,并對其進行測試[8],從宏觀方面解釋了一定量 Al的摻雜改善了LiMn2O4電池的循環性能,但是研究學者們對于鋁錳酸鋰的微觀結構研究較少[9]。本文根據密度泛函理論和第一性原理相結合對LMO結構模型進行一系列的分析和計算,對分別摻入Ni2+、Al3+前后LiMn2O4的總能量與晶格常數、總態密度的計算分析。通過對計算結果與實驗結論的對比,表明兩者的結論一致。
1 模型與方法
尖晶石LiMn2O4屬于立方晶系,為Fd-3m空間群[10],晶胞結構類型如圖1(a)所示。在尖晶石結構中,氧原子占據32e位置,錳原子占據16d位置,而鋰位于1/8氧四面體間隙(8a)位置[11]。Li+通過空的相鄰四面體和八面體間隙沿8a-16c-8a順序路徑在Mn2O4三維網絡結構中脫嵌和位置穿梭[12]。
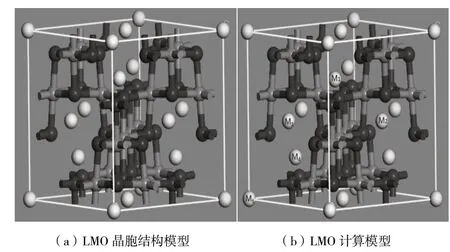
圖1 LMO晶胞體圖
它們沿傳導通道交替排列,并在整個晶體結構中建立了三維遷移網絡。一些具有較大四價陽離子的組合物,還顯示出由于Li+的置換而引起的較低對稱三斜晶相。在這種晶胞結構中,Li+在內部遷移按照8a-16c-8a順序移動[13],考慮到摻雜元素對晶體結構的變化及Li+的遷移,針對摻雜位對Li+遷移的影響,我們計算M1到M2方向的遷移,在圖1(b)中給出了在尖晶石中Li+的遷移路徑。觀察M1到M2的遷移過程中,摻雜元素對Li+的影響。
在晶體結構中離子遷移是由間隙或空位之間的離子熱活化跳躍驅動的。并且,宏觀擴散取決于材料的微觀結構。我們已經對LiMn2O4和相關材料的菱形結構的Li+擴散進行了大量實驗研究。在原子模型中研究運用密度泛函理論(DET)和動力學模擬研究LiMn2O4和LiNi0.5Mn1.5O4中的Li+遷移途徑。Vi+離子的部分取代針對多種元素進行,盡管用Ge等代替Ni取代Mn元素的結構確實穩定了金屬Li,但它們的生產成本明顯更高,并且存在具有較大陽離子的結構中的多態性問題。因此,與未取代的LMO相比,DFT研究集中于陽離子取代對晶體結構和能壘的影響。在以下各節中,描述了尖晶石結構模型和計算DFT設置,給出并討論了結果,最后給出了結論。
(1)LMO的結構模型。為了進行計算,使用了如圖1(a)所示的具有56個原子的尖晶石結構的常規六面體晶胞。除去一個Li原子以產生擴散所需的空位。作為參考結構,選擇了未取代的LMO組合物,對其余兩種陽離子(X=Ni,Al)取代Mn的LXMO組成進行了計算。對于每種組合物進行了晶胞體積和原子位置的弛豫。通過用晶胞LXMO中的X陽離子僅置換16個Mn原子之一,已經描述了較低濃度的取代元素對擴散路徑的直接影響。對于這些結構,僅原子位置已被弛豫,LMO的弛豫單元常數也已用于所有的LXMO,因為通過僅替換晶胞中的16個Mn原子之一,體積變化很小。
(2)幾何結構優化。本文采用Materials Studio軟件,在CASTEP中的Geometry Optimization模塊進行建模與能量計算,該模塊基于密度泛函理論對LMO與LXMO晶胞的幾何結構進行研究,計算中采用廣義梯度近似(GGA-PBE) 方案來描述電子與電子間的交換關聯能[14]。
(3)鋰空位和空位擴散。通過對Li+分布和晶體結構的對稱性的分析,我們得出了晶格中兩個獨立的鋰擴散路徑,它們將不同平面中的Li位點相互連接,并且因此,尖晶石結構中形成三維擴散網絡。然后通過NEB方法優化Li+遷移路徑,并且計算出遷移能壘。通過中子衍射(ND)實驗推導了LMO中Li優先占據M1位。LMO在溫室下顯示出Li陽離子原理M1位的無序現象,無法用K=0時的DFT計算來描述。首先假定空位介質的Li擴散。
(4)計算方法。計算使用CASTEP中的Energy計算模塊進行理論計算,計算參數設定為:原子間相互作用力的收斂標準設置為0.02eV/Ang,平面波截止為500eV,設置網格參數為4×4×4。原子最大位移收斂標準設置為1.0×10-5μeV。優化后數值與原始實驗設定晶胞常數a相比存在誤差,實際誤差為1.8%。通過使總能量最小化來完成晶胞體積的松弛,并且使原子位置松弛直至作用在原子上的剩余力小于10-3eV/。通過微動彈性帶(NEB)方法獲得相鄰M1個位點之間鋰躍遷的最小能量路徑(MEP)。作用于插值反應路徑的NEB圖像上的總力閾值設置為0.05eV/。
2 分析與討論
2.1 結構優化與總能量
根據總能量最低原理對三種模型優化處理,得到如表1所示優化后模型晶格參數和總能量。為了便于計算,節省計算時間,選用5×5×5模型進行計算。從圖 2(a)、(b)、(c)三種晶胞結構元素鍵可以看出它們都是Fd-3m對稱性的立方晶體。與LMO晶格常數初始值(a=8.221(4))對比,LMO結構優化后,原胞體積V=0.5609nm3,晶格常數減小,誤差僅為1.56%,在贗勢平面波方法所允許的范圍內,說明模型建立正確、可靠。與未摻雜相比,在Mn位摻雜Ni2+后導致晶格常數增大,原胞體積增大為0.5641nm3,總能量的變化量為-1152eV。而Mn位摻雜Al3+后導致晶格常數減小,原胞體積減小為0.5578nm3,總能量的變化量為-538eV。
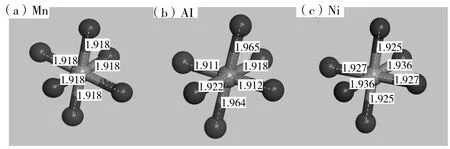
圖2 (a)LMO、(b)LAMO 和(c)LNMO 三種晶胞結構元素鍵
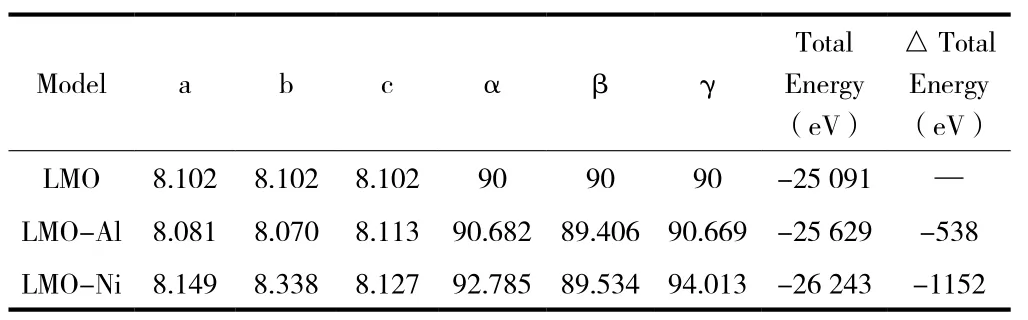
表1 優化后模型晶格常數和總能量
綜上所述,經過Mn位摻雜Ni2+、Al3+后體系能量的降低,說明晶體結構的穩定性提高。在Li+遷移時,結構不易會發生改變,能夠提高材料的循環穩定性和使用壽命。因此,在Mn位摻雜Ni2+、Al3+,均可以提高LMO結構的穩定性,且Mn位摻雜Ni2+的穩定性更好,與文獻[16]報道吻合。而Mn位摻雜元素的不同導致晶格常數變化不同,這可能與晶體結構、摻雜元素的濃度及摻雜位置有關。
2.2 總態密度與分態密度的分析
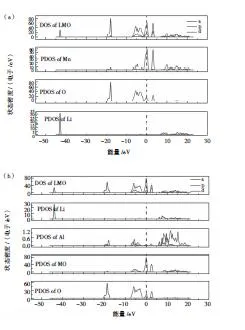
在第一性原理計算中,對幾種材料進行了晶胞結構優化。然后,分別得到圖3LMO、LAMO和LNMO的總態密度和分態密度圖,橫坐標為能量,縱坐標為態密度。通過元素摻雜后,元素之間的分布較為接近。這表明摻雜元素后,LMO尖晶石結構未發生結構改變,保留的尖晶石結構的穩定性。從圖3可以看出,在元素摻雜后,Mn與O的電子軌道發生輕微變化,表明材料的金屬性能未發生改變。
通過分析圖3可知,未摻雜與分別摻雜Al3+、Ni2+的三種材料在-7~1eV,隨著元素的替換,LAMO費米能級附近的峰主要由Al-p、O-s與Mn-d電子軌道進行貢獻,Mn、O元素的電子軌跡明顯重疊,由此說明Mn和Ni原子的加入,使得Mn與O之間的電子軌道明顯重疊,并且元素的替換,提高了元素間的相互作用,穩定了晶胞結構。在-20eV附近的電子軌道密度峰十分尖銳,說明成鍵作用較小,反鍵作用強烈,局域化明顯。由于Al的半徑小于Mn,原始Mn-O的鍵長縮短,這有利于提升Mn-O骨架的結構穩定性。在-40eV附近觀察看來,主要由Li-1s軌道提供,該峰的面積積分為2,表明該峰完全由Li-1s電子軌道中的兩個電子提供,該峰比較尖銳,且總態密度在費米能附近的峰強無Li的貢獻,說明Li與Mn-O作用很弱,Li可以在Mn-O結構中自由獨立地穿梭脫嵌,元素的摻雜導致材料性能提升。在Ni元素摻雜LMO晶胞結構后,使結構出現了O-2p能帶,導致在摻雜位出現Mn與O元素在電子軌道上的成鍵能力增強,這與文獻[17]的實驗結果一致。
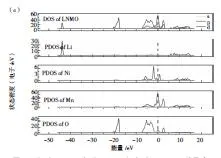
圖3 (a)LMO、(b)LAMO和(c)LNMO三種晶胞結構能量分布圖
2.3 晶胞結構的體相遷移
在晶胞結構圖中有三條遷移路徑由圖1(b)所示,分別為 M1-M2、M1-M3、M4-M5,對于遷移路徑的選擇,僅考慮摻雜元素對Li+遷移的影響,一方面是Li+的遷移通道,另一方面是局部的勢能面,這兩個因素相競爭,所以最終決定在摻雜位附近選取M1-M2兩個位置的Li+遷移情況進行理論計算。分別計算出M1-M2兩個位點的對向遷移能壘,得出遷移能壘相同,為0.74eV。之后首先計算出未摻雜位置錳原子的遷移能壘對Li+遷移的影響,在圖4(a)中可以看到在LMO晶胞結構中M1-M2遷移路徑,然后建立兩個晶胞結構,命名為生成物與反應物,接著計算間隙遷移需要的遷移能壘為0.72eV。在圖4(b)、(c)中我們可以看到經過Al與Ni原子替換后LMO晶胞結構中M1-M2的遷移路徑,對M1-M2兩個位置遷移都需要的能量,經過計算得出結果分別為0.32eV和0.57eV。由此得出結論:替換后的原子使得附近Li+遷移更加容易,Li+在Mn原子束縛下,距離近時受到Mn原子的影響比較大,而Mn原子經過Ni與Al原子替換后,鍵長變長,使得晶格變大,對Li影響較小,使其容易在晶胞結構中穿梭。
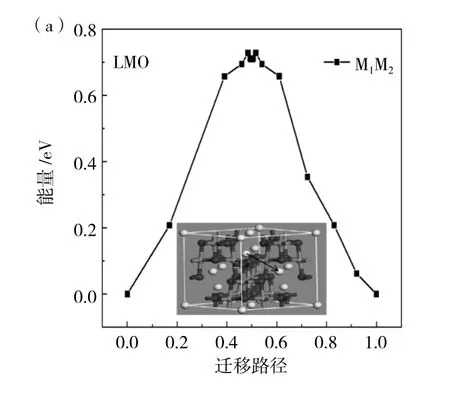
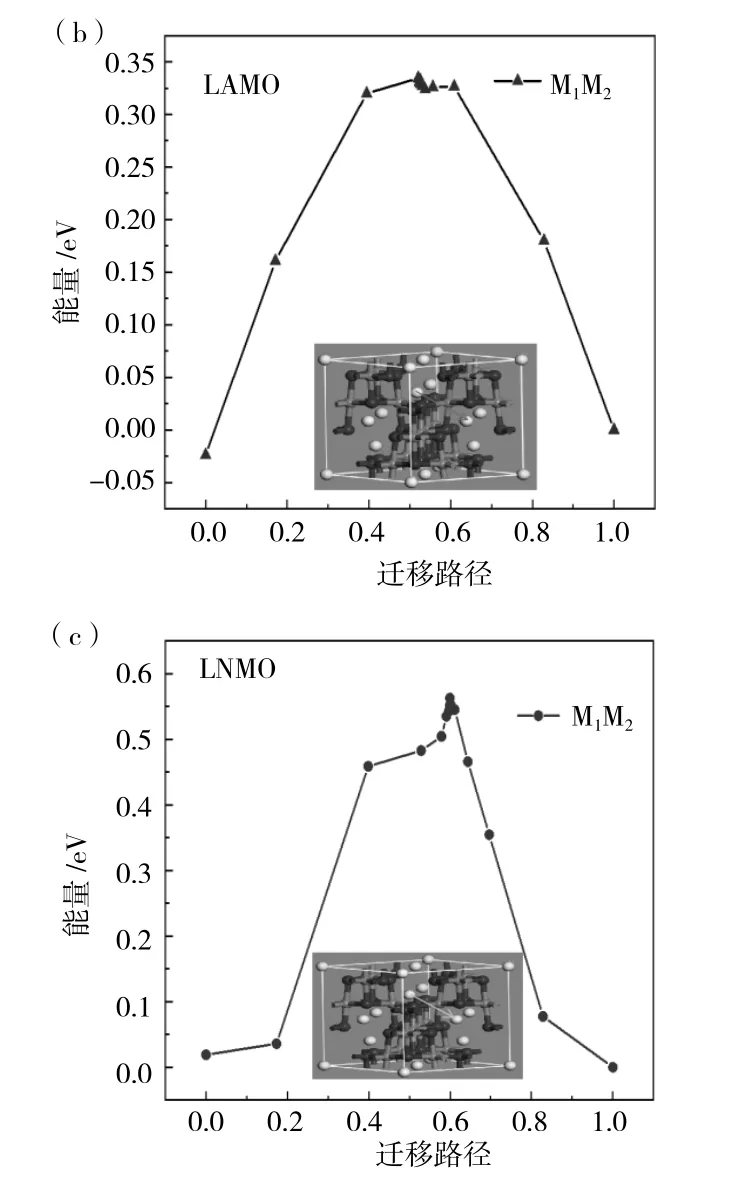
圖4 (a)LMO、(b)LAMO和(c)LNMO三種材料的遷移路徑能壘圖
3 結論
本文根據密度泛函理論以及第一性原理相結合對LMO結構模型進行一系列的分析和計算。通過結構分析可以看出,Mn位摻雜Ni2+、Al3+后LMO體系結構更穩定,且Mn位摻雜Ni2+時總能量的變化量為-1152 eV,穩定性更高。由態密度圖可以看出,在Mn位摻雜Al3+后,由于原始Mn-O的鍵長縮短,這有利于提升Mn-O骨架的結構穩定性,同時加快了LAMO晶胞結構中Li+的遷移速率。在LMO晶胞結構中,經過Ni元素替換Mn元素,由于嵌入了Ni-3d電子軌道,誘導LNMO增加了O元素的2p電子軌道,由于晶胞結構中Li+遷移所需要的能量大部分由O-2p能帶結構提供,因此,增加了尖晶石結構的導電性能。通過元素摻雜,使摻雜位鍵長變大。通過元素摻雜后,計算摻雜位置的鋰位遷移,計算出遷移能壘,由計算可知,經過元素替代Mn2+離子,摻雜后的過渡態能量能量降低,遷移勢壘更小。
猜你喜歡
小獼猴智力畫刊(2023年4期)2023-04-23 08:49:58
哲學評論(2021年2期)2021-08-22 01:53:34
中華詩詞(2019年7期)2019-11-25 01:43:04
模具制造(2019年3期)2019-06-06 02:10:54
中學生數理化·高一版(2018年1期)2018-02-10 05:20:03
影視與戲劇評論(2016年0期)2016-11-23 05:26:01
七彩語文·寫字與書法(2016年7期)2016-07-28 21:40:22
七彩語文·寫字與書法(2016年6期)2016-07-15 19:36:34
人間(2015年21期)2015-03-11 15:23:21
現代企業(2015年9期)2015-02-28 18:56:50
