巴洛沙韋關鍵中間體的合成工藝改進
2022-01-23 05:30:25于立國孫光祥張云然陶維潔
上海醫藥 2022年1期
于立國 孫光祥 張云然 陶維潔
摘 要 本研究報道了一條巴洛沙韋關鍵中間體3-(芐氧基)-4-氧代-4H-吡喃-2-羧酸(1)的新合成路線。以麥芽醇作為原料,經過芐基保護、縮合、氧化制得1。我們設計先將甲基間接轉變成烯胺,再將烯氨氧化成醛,與甲基直接氧化成醛相比,避免使用劇毒品二氧化硒,消除了對環境的污染,并且有效提高路線的安全性。同時,本研究對烯胺的氧化條件進行了選擇與優化,先將烯胺氧化成醛,醛中間體無需分離,進一步氧化到羧酸,“一鍋法”實現了兩個氧化歷程,并且避免使用昂貴的釕催化劑,大幅度降低成本。總收率69.7%,產品純度99.26%。
關鍵詞 巴洛沙韋 產業化 合成
中圖分類號:O625.8 文獻標志碼:A 文章編號:1006-1533(2022)01-0067-03
Improvement of synthetic process for key baloxavir intermediate
YU Liguo, SUN Guangxiang, ZHANG Yunran, TAO Weijie
(Changzhou Pharmaceutical Factory, Shanghai Pharmaceutical Group, Changzhou 213018, China)
ABSTRACT We reported a new synthetic route for baloxavir’s key intermediate 3-(benzyloxy)-4-oxo-4H-pyran-2-carboxylic acid (1). Maltol was used as the material and 1 was prepared by benzyl protection, condensation and oxidation. We designed the indirect conversion of methyl into enamine and then the oxidation of enamine into aldehyde. Compared with the direct oxidation of methyl into aldehyde, the new rout could avoid the use of selenium dioxide, eliminate the pollution to the environment and effectively improve the safety of the synthesis. Meanwhile, the oxidation conditions of enamine were selected and optimized in this study. The enamine was oxidized to aldehyde first, and the aldehyde intermediate was further oxidized to carboxylic acid without separation. The “one-pot method” realized two oxidation processes and avoided the use of expensive ruthenium catalyst, which could greatly reduce the cost. The total yield was 69.7% and the purity was 99.26%.
KEy wORDS baloxavir; industrialization; synthesis
巴洛沙韋(baloxavir,商品名Xofluza)是一種首創(first-in-class)、單劑量口服藥物,具有不同于市面其他抗病毒藥物的全新抗流感作用機制[1],是一種對流感病毒的復制必不可少的內切核酸酶抑制劑。巴洛沙韋具有非常好的市場潛力,與2009年銷售額達32億美元的競爭品種羅氏公司的奧司他韋相比[2],具有更快的抗病毒療效,服藥方案簡單,不良反應更小,有望成為繼奧司他韋后繼續領跑抗流感藥的王牌之一。
文獻報道的3-(芐氧基)-4-氧代-4H-吡喃-2-羧酸(1)的合成路線主要有兩條。①以麥芽醇為起始原料,經過芐基保護,制得3-(芐氧基)-2-甲基-4H-吡喃-4-酮(2),2經過二氧化硒氧化,制得3-(芐氧基)-4-氧基-4H-吡喃-2-甲醛(3),3經過亞氯酸鈉氧化制得1[3]。其環境污染嚴重(須使用劇毒品二氧化硒[4]),反應溫度高(155℃),收率較低,不適合工業化生產。②以麥芽醇為起始原料,經過芐基保護,制得2,2用LiHMDS拔氫后與苯甲醛發生縮合反應,制得3-(芐氧基)-2-(2-羥基-2-苯乙基)-4H-吡喃-4-酮(4),4與甲磺酰氯取代生成活性甲磺酸酯,在DBU存在的情況下發生脫水反應,生成(E)-3-(芐氧基)-2-苯乙烯基-4H-吡喃-4-酮(5),5在三氯化釕和高碘酸鈉的條件下發生氧化反應,生成3,3經過亞氯酸鈉氧化制得1[5]。其合成路線長,成本高(須使用釕催化劑[6]),加之環境污染嚴重,也不適合工業化生產。
1是合成巴洛沙韋的關鍵中間體,鑒于巴洛沙韋巨大的藥用前景,開發一條環境友好、成本低、適合產業化的合成路線變得尤為重要。
1 材料與方法
1.1 儀器
LC-20AD高效液相色譜儀(日本島津公司);ZF-8型暗箱式四用紫外線分析儀(上海嘉鵬科技有限公司);GF254薄層層析(TLC)板、P6107-C電子天平(阿拉丁公司)。
1.2 試劑
麥芽醇、N,N-二甲基甲酰胺二甲縮醛(DMF-DMA)及其他試劑均為市售工業級商品。
1.3 方法
1.3.1 關鍵中間體1合成路線
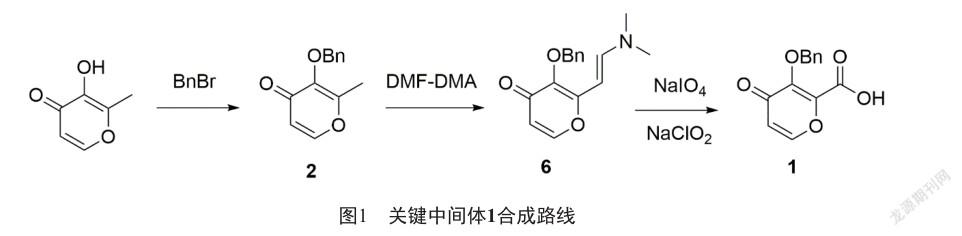
我們開發了一條環境友好、成本低、適合產業化的新合成路線(圖1)。該路線以廉價易得的麥芽醇為起始原料,經過芐基保護,制得2,2與N,N-二甲基甲酰胺二甲基縮醛縮合生成(E)-3-(芐氧基)-2-(2-(二甲氨基)乙烯基)-4H-吡喃-4-酮(6),6經過高碘酸鈉[7]和亞氯酸鈉氧化[8]制得1。
1.3.2 HPLC檢測
采用峰面積歸一化法:Waters SunFire C18柱(4.6 mm×150 mm, 5mm);流動相 A 為0.01 mol/L Na2HPO4-三乙胺(TEA)=100∶0.1(H3PO4調pH=6.0)、B為甲醇,梯度洗脫(0~20 min:A 80%;20~25 min:A 50%;25~45 min:A 10%);柱溫35 ℃;流速1.0 mL/min;檢測波長225 nm;進樣量10 mL;保留時間 22.6 min。
2 結果
2.1 化合物2的制備
將麥芽醇(1 890 g,14.99 mol)溶于18.9 L N,N-二甲基甲酰胺,加入溴化芐(1.84 L, 15.49 mol),碳酸鉀(2 280 g,16.50 mol),加熱到80 ℃,保溫2 h,薄層色譜法檢測原料完全轉化,降溫至室溫,加入10 L四氫呋喃,抽濾,濾液于45 ℃減壓濃縮至干得到白色固體(3 290 g,15.21 mol),收率100%。
1H NMR(400 MHz, CDCl3)δ 7.59(d,J=5.6 Hz, 1H)、7.42~7.29(m,5H)、6.37(d,J=5.6 Hz,1H)、5.16(s,2H)、2.08(s,3H)。
2.2 化合物6的制備
將化合物2(1 080 g,4.99 mol)溶于5.4 L N,N-二甲基甲酰胺,加入N,N-二甲基甲酰胺二甲基縮醛(1 480 g,12.42 mol),加熱至回流,保溫8 h,薄層色譜法檢測原料完全轉化,降溫至室溫,反應液中加入40 L水,16 L二氯甲烷萃取3次,合并有機層,棄去水層,有機層用適量無水硫酸鈉干燥,抽濾,濾液于30 ℃減壓濃縮至干,得到油狀物(1 153 g,4.25 mol),收率85%。
1H NMR(400 MHz,CDCl3) δ 7.45(d,J=6.4 Hz,2H)、7.41(d,J=5.6 Hz,1H)、7.36~7.27(m,3H)、7.02(d,J=13.4 Hz,1H)、6.21(d,J=5.6 Hz,1H)、5.10(s,2H)、5.00(d,J=13.4 Hz,1H)、2.83(s,6H)。
2.3 化合物1的制備
將化合物6(1 466 g,5.40 mol)溶于7 L N,N-二甲基甲酰胺,加入高碘酸鈉(2 310 g,10.80 mol)的15 L水溶液中,過程中控制反應液溫度不超過25 ℃,加畢,反應液室溫攪拌1 h,薄層色譜法檢測原料完全轉化,抽濾,濾餅用2 L乙酸乙酯漂洗,合并濾液,濾液用10 L乙酸乙酯萃取3次,合并有機層,有機層用5 L飽和氯化鈉洗滌三次,有機層用無水硫酸鈉干燥,抽濾,濾液濃縮得到油狀物。將亞氯酸鈉(1 466 g,16.21 mol)和氨基磺酸(1 052 g,10.84 mol)溶于7 L水中,將上述油狀物用7 L丙酮溶解,滴加到水溶液中,控制溫度不超過25 ℃,保溫1 h,薄層色譜法檢測原料完全轉化,將反應液濃縮除去丙酮,析出大量白色固體,降溫至5 ℃,抽濾,鼓風干燥,得到白色固體(1 092 g,4.44 mol),收率82%。HPLC檢測產品純度為99.26%。
1H NMR (400 MHz, DMSO) δ 14.23(s,1H)、8.19(d,J=5.6 Hz,1H)、7.42(d,J=6.7 Hz,2H)、7.36~7.29(m,3H)、6.54(d,J=5.6 Hz,1H)、5.10(s,2H)。
3 討論
本工藝與現有文獻方法相比,有如下優點:①先將甲基轉變成烯胺,再氧化成醛,避免使用劇毒的二氧化硒,大幅減少環境污染;同時顯著提高氧化的收率,有效降低了成本。②構建烯胺僅一步反應,與構建碳碳雙鍵相比,既避免強堿低溫給車間操作帶來的麻煩,又避免使用劇毒品甲磺酰氯,綠色環保,大幅縮短反應步驟,收率很高,有利于成本控制。③通過對反應條件的優化,實現“一鍋法”將烯胺氧化到羧酸,與碳碳雙鍵的氧化相比,無需使用昂貴的三氯化釕,且反應收率更高。
改進后的工藝,反應條件溫和、操作簡便、手性純度高、成本低廉,總收率69.7%,產品純度99.26%,適合工業化生產。
致謝:感謝浙江大學為本研究的核磁共振檢測提供支持。
參考文獻
[1] 李佳悅, 劉洋. 巴洛沙韋[J] 中國藥物化學雜志, 2019, 29(5): 411.
[2] 李秋, 王珊. 抗病毒藥物的研究進展[J]. 醫藥導報, 2011, 30(6): 732-735.
[3] Puerta DT, Botta M, Jocher CJ, et al. Tris(pyrone) chelates of Gd(Ⅲ) as high solubility MRI-CA[J]. J Am Chem Soc, 2006, 128(7): 2222-2223.
[4] 藍柳恒. 二氧化硒生產過程職業病危害的預防及控制[J].中國衛生產業, 2015, 12(11): 6-8.
[5] Aoyama Y, Hakogi T, Fukui Y, et al. Practical and scalable synthetic method for preparation of dolutegravir sodium: improvement of a synthetic route for large-scale synthesis[J]. Org Process Res Dev, 2019, 23(4): 565-570.
[6] 龍思, 宇裴響, 林羅丹, 等. 貴金屬釕基催化劑合成及應用研究進展[J]. 化學通報, 2021, 84(2): 120-128.
[7] Bernd P. Selectivity versus reactivity-recent advances in RuO4-catalyzed oxidations[J]. Synthesis, 2005(15): 2453-2472.
[8] 姜志猛. 亞氯酸鈉體系選擇性氧化制備布洛芬及其它羧酸[J]. 中國醫藥工業雜志, 1991, 22(1): 1-2.
