UPLC-MS/MS法測定止咳平喘中藥制劑中16種非法添加化學藥物
2022-01-27 14:41:38汪瑾彥
中成藥 2022年1期
關鍵詞:檢測
汪瑾彥, 梁 祈, 莊 玥, 蔡 霞
(廣東省藥品檢驗所,廣東 廣州 510663)
隨著經濟社會發展,哮喘已成為常見疾病之一[1]。一些不法分子為了達到速效目的,在止咳平喘中藥制劑中非法添加化學藥物,患者在不知情時服用了這些藥物,可能會對健康造成嚴重危害。
目前,我國止咳平喘類中藥中非法添加化學藥物的檢測標準是藥品補充檢驗方法2009031,涉及8種藥物(茶堿、磺胺甲噁唑、醋酸潑尼松、地西泮、馬來酸氯苯那敏、鹽酸苯海拉明、枸櫞酸噴托維林、磷酸苯并哌林),采用HPLC法檢查和定量,LC-MS/MS法確證,并且均各采用2個色譜系統進行分離。但該類添加通常為復合添加,故亟需提高檢驗效率,擴充檢驗成分。
上述藥物大多采用HPLC法[2-5]、LC-MS/MS法[6-11]、薄層色譜法[12-14]等技術,通常以檢測8~18種為主[6-11],一般檢出1~4種陽性[6-10],部分側重于定性篩查[9-10]。本研究選擇藥品補充檢驗方法2009031中的8種藥物,結合近年來發現的其他8種作為研究對象,建立UPLC-MS/MS法同時測定上述16種非法添加物的檢測方法,以期為相關監管提供有力的技術支撐。
1 材料
1.1 儀器 LC-30 超高效液相色譜儀(日本島津公司);SCIEX QTRAP5500三重四極桿線性離子阱質譜儀(配ESI源)(美國Sciex公司);KQ-300DE超聲波清洗器(昆山市超聲儀器有限公司)。
1.2 試藥 茶堿、磺胺甲噁唑、醋酸潑尼松、地西泮、馬來酸氯苯那敏、鹽酸苯海拉明、枸櫞酸噴托維林、磷酸苯并哌林、對乙酰氨基酚、磷酸可待因、甲氧芐啶、鹽酸二氧丙嗪、氫溴酸右美沙芬、阿司匹林、雙氯芬酸鈉對照品均購自中國食品藥品檢定研究院;鹽酸異丙嗪對照品購自德國Dr.Ehrenstorfer公司。乙腈、甲酸(色譜純);甲醇、甲酸銨(分析純);0.22 μm針式濾膜過濾頭(津騰實驗室設備有限公司);實驗用水為超純水。
2 方法
2.1 對照品溶液制備 精密稱取上述16種藥物對照品各10 mg,置于同一10 mL棕色量瓶中,甲醇溶解定容至刻度,制成質量濃度為1.0 mg/mL的貯備液,甲醇逐級稀釋成1.0、2.0、3.0、5.0、10.0、25.0、50.0、100.0 ng/mL系列溶液,即得。
2.2 樣品前處理
2.2.1 固體樣品 膠囊取內容物,片劑、丸劑研磨成粉末后混勻,準確稱取0.3 g至50 mL量瓶中,加適量甲醇超聲提取15 min,放冷至室溫,甲醇定容至刻度,搖勻,根據實際質量濃度稀釋至線性范圍內,過0.22 μm微孔濾膜后待測。
2.2.2 液體制劑 搖勻后,準確量取1.0 mL至50 mL量瓶中,加甲醇適量,超聲提取15 min,放至室溫,甲醇定容至刻度,搖勻,根據實際濃度稀釋至線性范圍內,過0.22 μm微孔濾膜后待測。
2.3 分析條件
2.3.1 色譜 Waters ACQUITY UPLC BEH C18色譜柱(2.1 mm×100 mm,1.7 μm);流動相乙腈(A)-10 mmol/L甲酸銨(0.1%甲酸)(B),梯度洗脫(0~0.5 min,5%A;0.5~9 min,5%A~70%A;9~10 min,70%A;10~11 min,70%A~95%A;11~13 min,95%A;13~13.1 min,95%A~5%A;13.1~16 min,5%A);體積流量0.3 mL/min;柱溫40 ℃;樣品室溫度4 ℃;進樣量1 μL。
2.3.2 質譜 電噴霧離子源(ESI),多反應監測(MRM);氣簾氣35 psi(1 psi=6.895 kPa);離子源溫度550 ℃;噴霧氣55 psi;輔助加熱氣55 psi;阿司匹林、雙氯芬酸為負離子模式掃描,離子化電壓-4 500 V;其他藥物為正離子模式掃描,離子化電壓5 500 V。質譜參數見表1。

表1 16種藥物優化質譜條件
3 結果與討論
3.1 色譜條件優化
3.1.1 色譜柱選擇 本實驗比較了Waters ACQUITY UPLC BEH C18(2.1 mm×100 mm,1.7 μm)、Waters ACQUITY UPLC HSS T3色譜柱(2.1 mm×50 mm, 1.8 μm),結果前者分離效果較后者更優,并且可滿足儀器分析要求。
3.1.2 流動相選擇 止咳平喘類藥物通常以有機相(乙腈或甲醇)-緩沖鹽水溶液的混合溶劑體系作為流動相,由于乙腈柱壓更小,而且質譜響應值更高,故選用乙腈作為有機相[15]。
本實驗分別以乙腈-0.1%甲酸、乙腈-20 mmol/L乙酸銨、乙腈-10 mmol/L甲酸銨(0.1%甲酸)、乙腈-10 mmol/L乙酸銨(0.1%乙酸)為流動相,結果乙腈-0.1%甲酸、乙腈-20 mmol/L乙酸銨洗脫時,部分目標物保留時間接近,峰型較差;乙腈-10 mmol/L甲酸銨(0.1%甲酸)、乙腈-10 mmol/L乙酸銨(0.1%乙酸)洗脫時,目標物響應值較高,峰型較好,大部分藥物能較好地分離。考慮10 mmol/L甲酸銨(0.1%甲酸)體系中地西泮與雙氯芬酸分離度優于10 mmol/L乙酸銨(0.1%甲酸)體系中,因此,選擇乙腈-10 mmol/L甲酸銨(0.1%甲酸)作為流動相。
3.2 質譜條件優化 本實驗采用直接進樣的方式,分別將16種藥物對照品溶液注入質譜的離子源中,優化質譜條件,分別在正、負離子模式下進行Q1全掃描,選擇合適的電離方式和準分子離子。再對準分子離子進行MS2全掃描,以1~2個相對豐度最高的子離子作為定性離子,其中相對豐度較高的離子作為定量離子,然后,在多反應監測模式(MRM)下優化各子離子的去簇電壓(DP)和碰撞能(CE),MRM色譜圖見圖1。
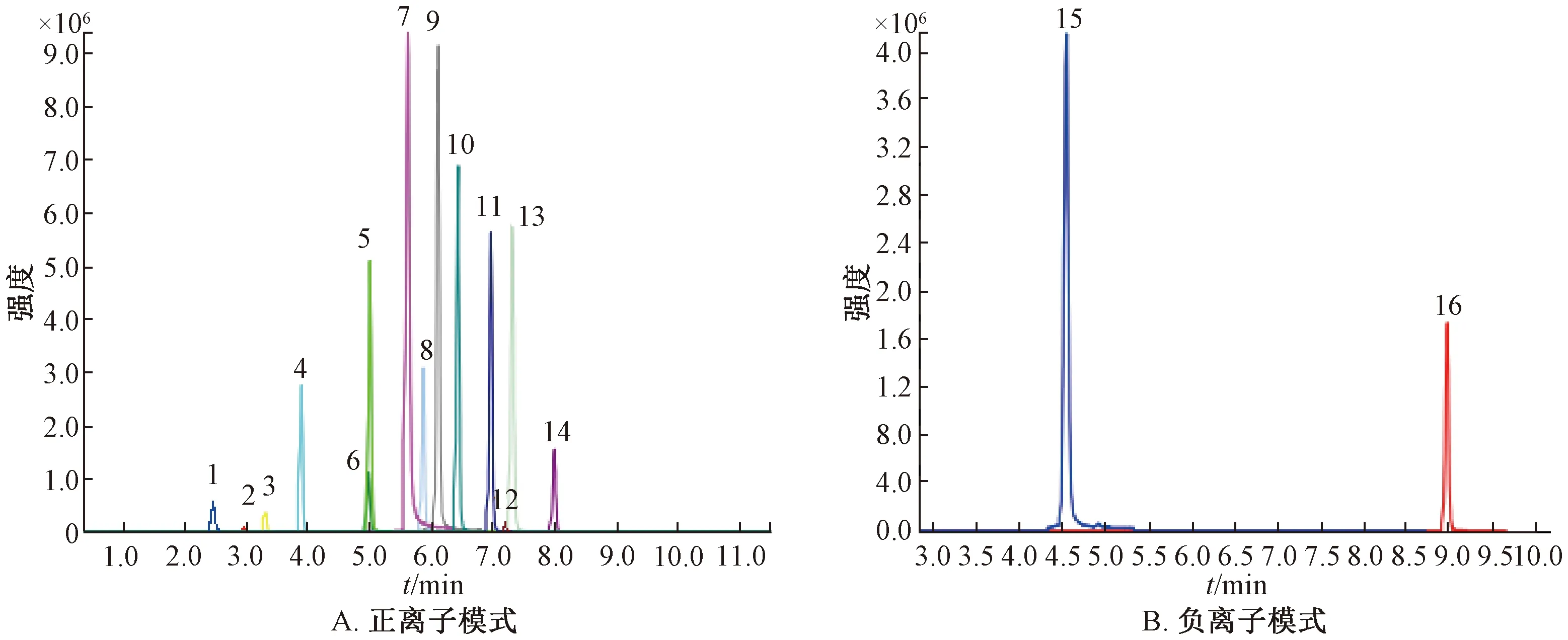
1.對乙酰氨基酚 2.茶堿 3.可待因 4.甲氧芐啶 5.鹽酸二氧丙嗪 6.磺胺甲噁唑 7.馬來酸氯苯那敏8.氫溴酸右美沙芬 9.鹽酸苯海拉明 10.鹽酸異丙嗪 11.枸櫞酸噴托維林 12.醋酸潑尼松 13.磷酸苯丙哌林14.地西泮 15.阿司匹林 16.雙氯芬酸鈉1.paracetamol 2.theophylline 3.codeine 4.trimethoprim 5.dioxopromethazine hydrochloride 6.sulfamethoxazole 7.chlorpheniramine maleate 8.dextromethorphan hydrobromide 9.diphenhydramine hydrochloride 10. promethazine hydrochloride 11.pentoxyverine citrate 12.prednisone acetate 13.benproperine phosphate 14.diazepan 15.aspirin 16.diclofenac sodiom圖1 各藥物MRM色譜圖Fig.1 MRM chromatograms of various drugs
3.3 提取溶劑選擇 止咳平喘類藥物一般以甲醇為溶劑[2-10],這可能是由于它們具有較好的脂溶性。本實驗考察了甲醇對16種藥物的提取效果,發現溶解性均較好,回收率達到檢測要求,綜合考慮成本、試劑毒性等因素,最終以其為提取溶劑。
3.4 方法學考察
3.4.1 專屬性試驗 本實驗選擇陽性檢出率較高的膠囊劑和口服液2種基質,分別作為固態和液態止咳平喘中藥制劑的代表基質。取膠囊和口服液陰性樣品若干批,按“1.3”項下方法進行前處理后進樣分析,結果均未檢出。
3.4.2 線性關系考察 本實驗在優化色譜-質譜條件下,對系列質量濃度對照品溶液進行測定,以MRM定量離子對峰面積為縱坐標(Y),質量濃度為橫坐標(X)進行回歸,結果見表2,表明各藥物在各自范圍內線性關系良好。再以定量離子對的信噪比S/N≥3作為LOD,≥10作為LOQ,結果見表2。
3.4.3 加樣回收率試驗 本實驗取上述2種基質的陰性樣品,加入對照品適量,制成3個質量濃度,每個水平6份,按“1.3”項下方法處理后進樣檢測,結果見表3。
3.4.4 穩定性試驗 取10 μg/L對照品溶液,于 0、2、4、8、12、18、24 h 進樣檢測,測得各成分峰面積RSD為2.4%~11.0%,表明溶液在24 h內穩定性良好。
3.5 陽性樣品檢測 從互聯網和實體經營場所收集42批樣品,按上述方法檢測,檢出陽性樣品13批(膠囊7批、粉劑3批、丸劑1批、液體制劑2批),檢出率31.0%,其中有10批來自網購,表明網絡銷售產品具有較大風險;共檢出12種藥物,其中茶堿、醋酸潑尼松、馬來酸氯苯那敏是最為常見的非法添加;較補充檢驗方法2009031新增的8種藥物均檢出陽性樣品,其中右美沙芬、異丙嗪為國內首次報道。具體見表4。
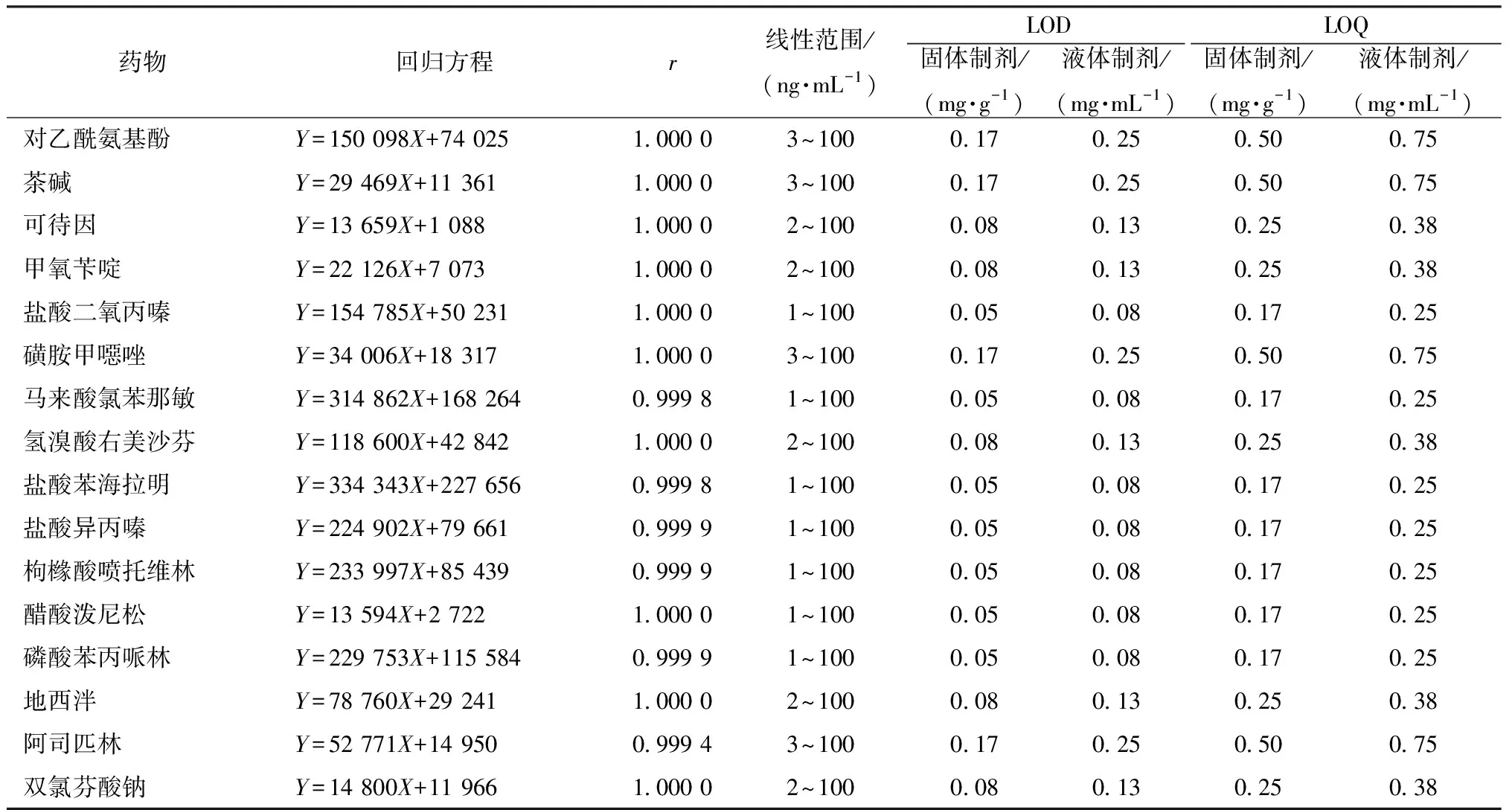
表2 各藥物線性關系Tab.2 Linear relationships of various drugs
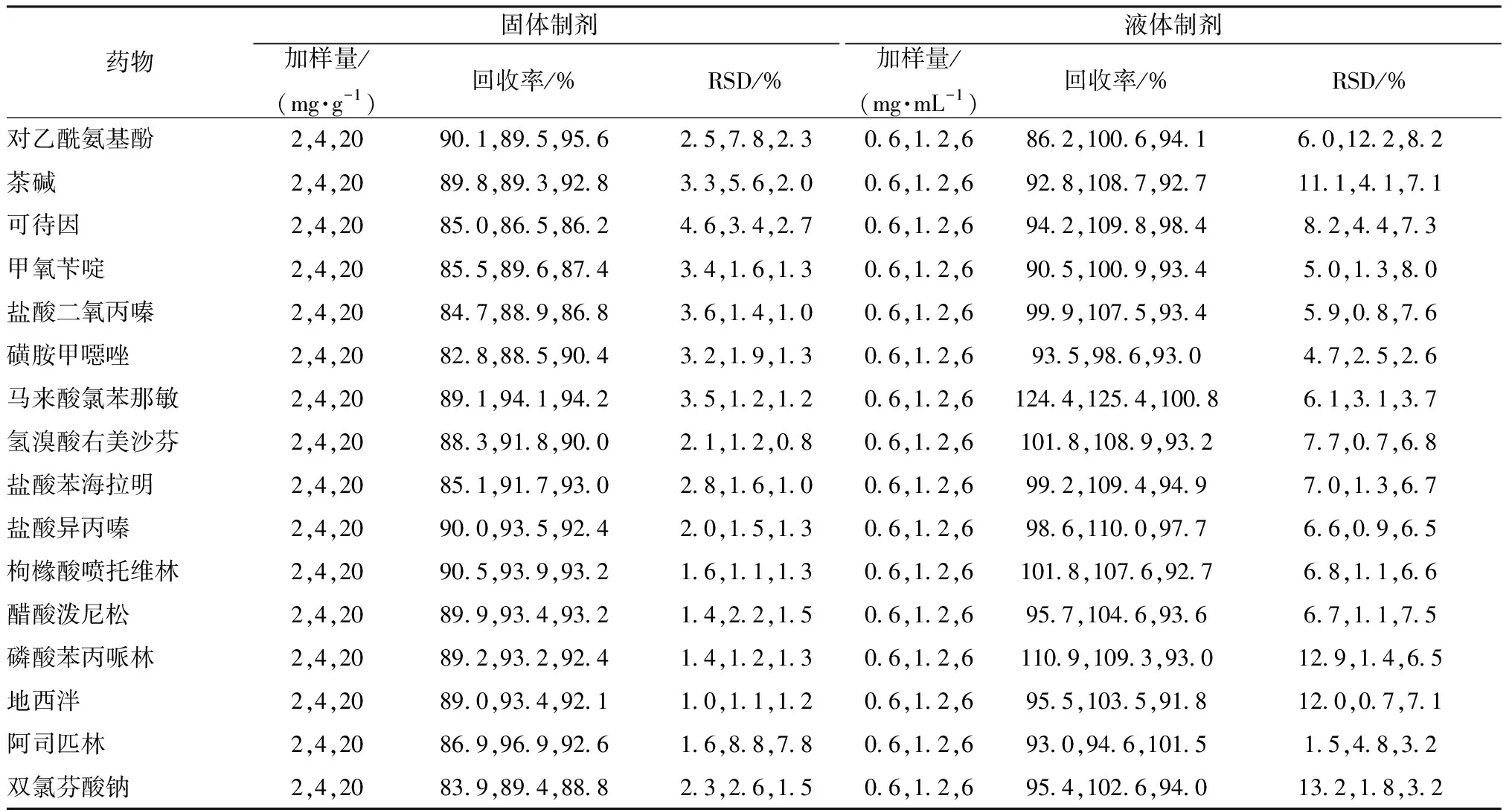
表3 各藥物加樣回收率試驗結果(n=6)
不合格樣品中,普遍存在多種藥物復合添加的情況,而且含量差異巨大,最常見的方式為“茶堿+醋酸潑尼松+其他”,這種聯合用藥的隨意性可能對患者健康造成嚴重的損害。
4 結論
本實驗建立UPLC-MS/MS法同時測定止咳平喘類中藥制劑中16種非法添加化學藥物,擴充了現有國家標準的檢驗藥物,結果均檢出陽性樣品,并且首次報道了氫溴酸右美沙芬和鹽酸異丙嗪,該方法實用性強,可為相關監督檢驗提供有力的技術支撐。

表4 陽性樣品檢測結果
猜你喜歡
中國設備工程(2022年12期)2022-07-11 04:33:00
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:36
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:34
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:50
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:48
