新疆地區光果甘草與脹果甘草化學成分差異的多元統計分析△
2022-01-28 13:28:52陳佳聶黎行戴勝云劉薇魏鋒馬雙成
中國現代中藥 2021年12期
關鍵詞:差異
陳佳,聶黎行,戴勝云,劉薇,魏鋒,馬雙成
中國食品藥品檢定研究院,北京 102629
甘草為豆科植物甘草Glycyrrhiza uralensisFisch.、脹果甘草G.inflataBat.或光果甘草G.glabraL.的干燥根和根莖,在我國有悠久的使用歷史,近60%的中藥成方制劑中含有甘草[1]。甘草具有補脾益氣、清熱解毒、祛痰止咳、緩急止痛、調和諸藥的功效[2],《神農本草經》將其列為上品,又名“國老”。據本草考證,歷代古籍所描述的甘草基原為甘草G.uralensisFisch.,多產于寧夏、內蒙古、陜西、甘肅一帶,以“梁外甘草”為最佳,即內蒙古鄂爾多斯杭錦旗梁外地區[3-5]。新疆地區甘草主要包括3 種基原,分別是甘草G.uralensisFisch.、脹果甘草G.inflataBat.和光果甘草G.glabraL.,其中脹果甘草系新疆地區特有的甘草藥用資源,主要分布于新疆的南疆塔里木盆地和東疆哈密、吐魯番地區及塔里木河及孔雀河流域等地區[6]。光果甘草主產于中東及地中海地區,在我國主要分布在新疆北部及甘肅等地區[7-8]。
本課題組前期研究對甘草G.uralensisFisch.化學成分進行了詳細分析[9],本實驗在前期研究基礎上,針對新疆地區光果甘草和脹果甘草化學成分進行分析和探討。目前關于脹果甘草和光果甘草化學成分的研究報道很多,大多針對其中1 種基原開展深入研究[10-11],也有相關報道對脹果甘草和光果甘草進行化學成分對比研究,如含量測定方法的建立[12]、三萜皂苷類或黃酮類化合物的含量分析[13-14]、不同基原甘草的化學成分差異探討[15]等。本實驗采用高效液相色譜法(HPLC)測定了光果甘草和脹果甘草不同結構類型的20個化學成分(2個三萜皂苷類、2個香豆素類、3 個查耳酮類、13 個黃酮類化合物)含量,并運用多元統計分析,找出光果甘草和脹果甘草之間的差異化學成分,為多基原甘草藥材質量評價及相關標準制定提供參考。
1 材料
1.1 儀器
Waters e2695 型高效液相色譜儀(2998 二極管陣列檢測器、Empower 網絡版工作站);KQ-250DE型醫用數控超聲波清洗器(昆山市超聲儀器有限公司);XSE 205DU 型電子天平(梅特勒-托利多儀器公司)。
1.2 試劑
對照品甘草酸銨(批號:110731-202021,純度:96.20%)、甘草苷(批號:111640-201908,純度:95.00%)、芒柄花素(批號:111703-201504,純度:95.00%)均來自中國食品藥品檢定研究院;甘草皂苷G2(批號:PCS200904,純度:98.59%)購自成都植標化純生物技術有限公司;甘草異黃酮B(批號:PS20110202,純度:98.02%)、甘草酚(批號:PS010089,純度:98.71%)均購自成都普思科技股份有限公司;半甘草異黃酮B(批號:MUST20072104,純度:99.91%)、甘草黃酮醇(批號:MUST20041311,純度:98.86%)均購自成都曼思特生物科技有限公司;芹糖甘草苷(批號:PRF9050224,純度:99.95%)、芹糖異甘草苷(批號:PRF9101021,純度:97.04%)、甘草素(批號:PRF20042742,純度:99.50%)、異甘草素(批號:PRF20060943,純度:99.87%)、光甘草酚(批號:PRF20032401,純度:99.95%)、刺甘草查耳酮(批號:PRF10122621,純度:99.81%)、異甘草苷(批號:PRF20040923,純度:98.23%)、新異甘草苷(批號:PRF20060942,純度:99.25%)、甘草香豆素(批號:PRF20060921,純度:99.77%)、甘草查耳酮A(批號:PRF20033022,純度:98.57%)、甘草查耳酮B(批號:PRF10101021,純度:99.74%)、佛來心苷(批號:PRF9110601,純度:95.77%)均購自成都普瑞法科技開發有限公司;甲酸、乙腈均為色譜純;純凈水為Milli-Q 超純水,其他試劑均為分析純。
10批脹果甘草(S1~S10)及10批光果甘草(S11~S20)樣品經中國食品藥品檢定研究院中藥標本館張南平主任藥師鑒定,分別為豆科植物光果甘草Glycyrrhiza glabraL.及脹果甘草G.inflataBat.的干燥根和根莖,見表1。樣品保存于中國食品藥品檢定研究院中藥民族藥檢定所中藥標本館。
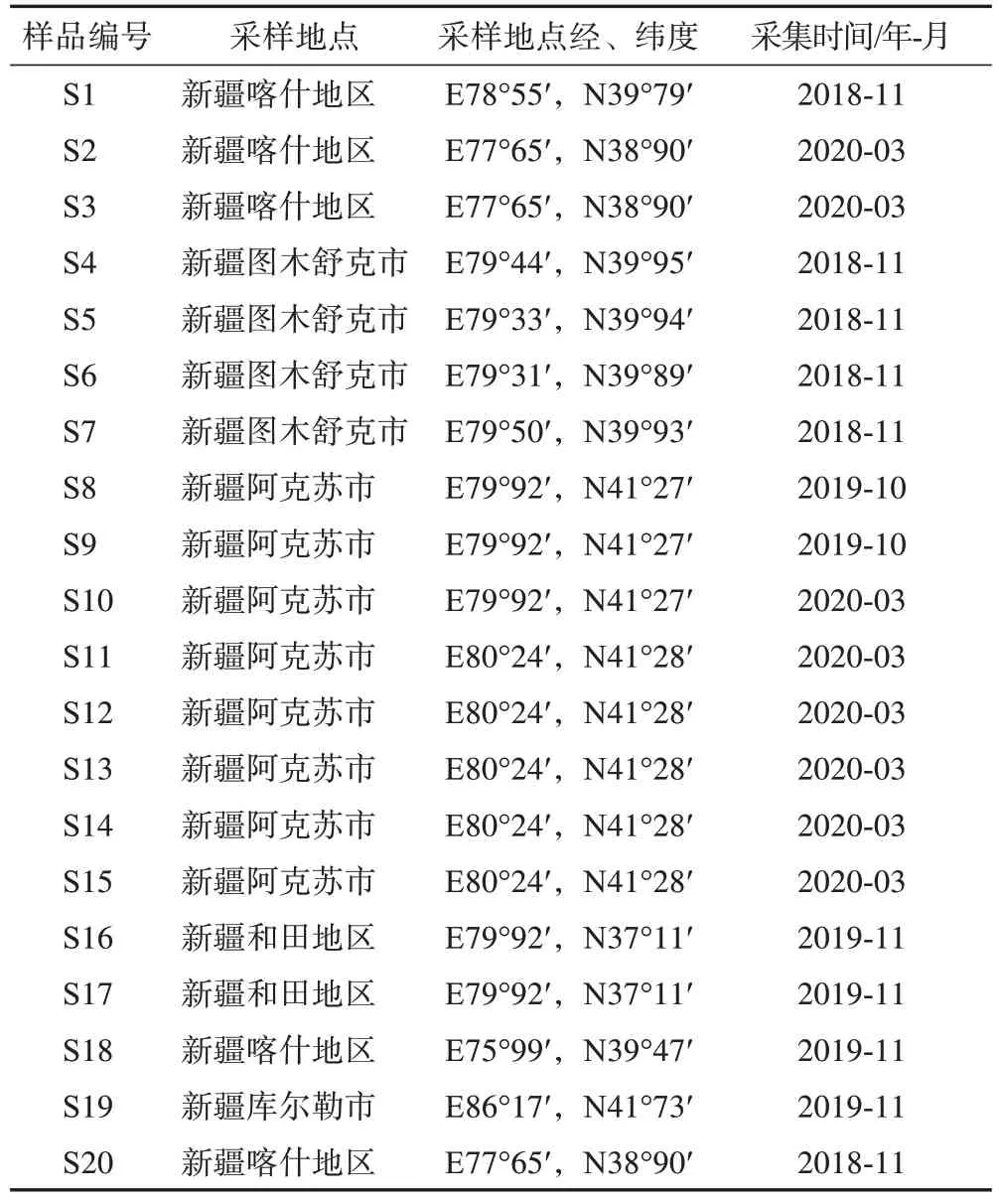
表1 光果甘草及脹果甘草藥材信息
2 方法
2.1 光果甘草與脹果甘草中16個化合物含量測定
2.1.1 色譜條件 Shiseido Capcell Pak MG C18色譜柱(250 mm×4.6 mm,5 μm),流動相為乙腈(A)-0.1%磷酸水溶液(B),梯度洗脫。甘草查耳酮B、刺甘草查耳酮、甘草查耳酮A 及光甘草酚洗脫梯度為:0~30 min,25%~70%A;30~34 min,70%A;34~35 min,70%~95%A;35~40 min,95%A;40~41 min,95%~25%A;41~45 min,25%A。其余16 個化合物洗脫梯度為0~60 min,5%~95%A;60~65 min,95%A;65.0~65.2 min,95%~5%A;65.2~75.0 min,5%A。流速為1 mL·min-1。柱溫為40 ℃。進樣量為10 μL。檢測波長為250 nm(芒柄花素、甘草酸和甘草皂苷G2)、262 nm(半甘草異黃酮B 和甘草異黃酮B)、275 nm(甘草苷、芹糖甘草苷和甘草素)、360 nm(佛來心苷、芹糖異甘草苷、異甘草苷、新異甘草苷、異甘草素、甘草香豆素、甘草酚、甘草黃酮醇、甘草查耳酮B、刺甘草查耳酮和甘草查耳酮A)、282 nm(光甘草酚)[9,16]。色譜圖見圖1~2。
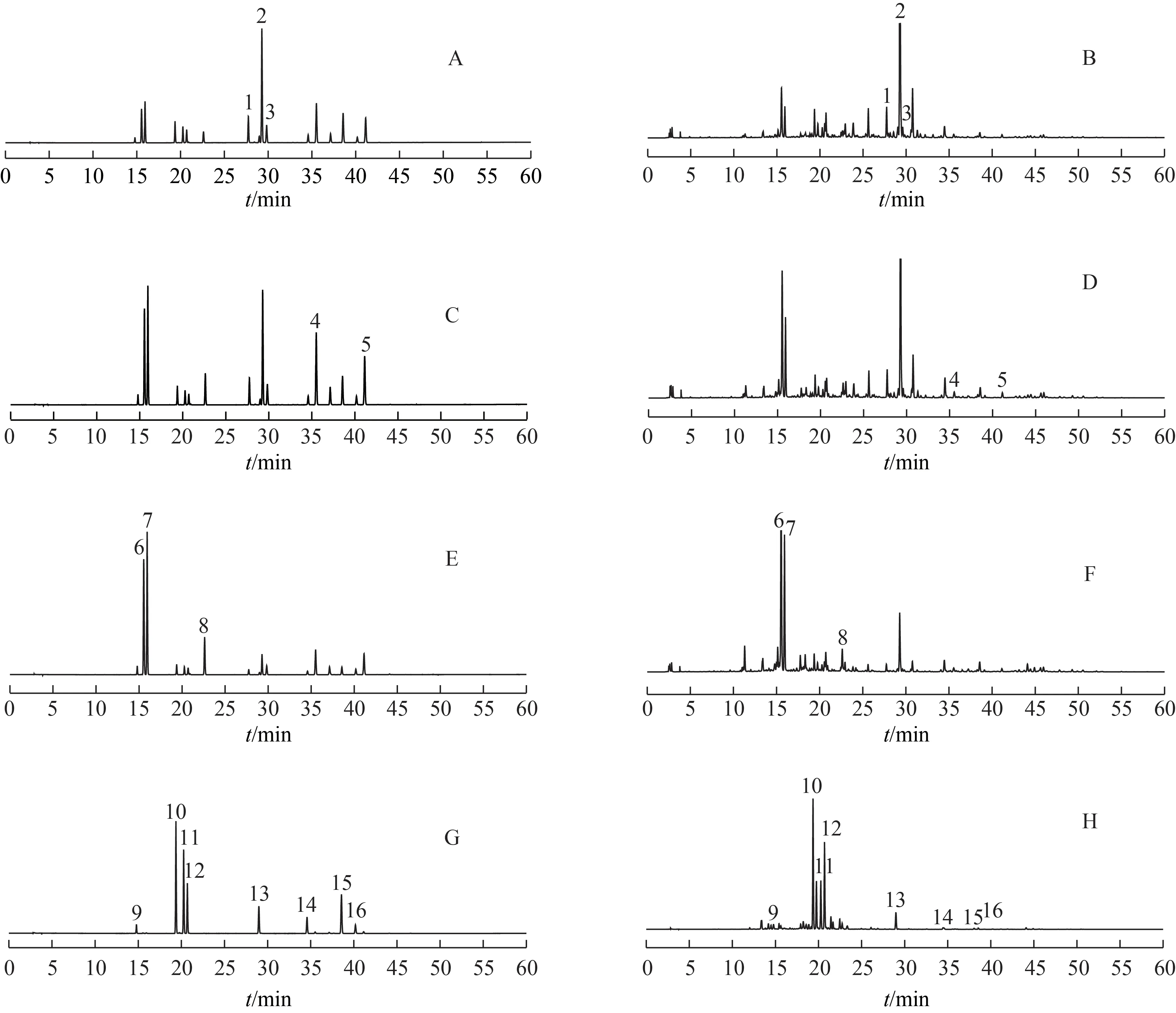
圖1 甘草藥材16個化學成分HPLC圖
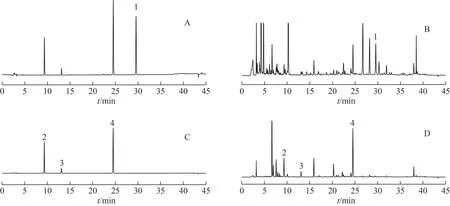
圖2 甘草藥材4個化學成分HPLC圖
2.1.2 對照品溶液的制備 精密稱取甘草皂苷G2、甘草酸銨、芒柄花素、半甘草異黃酮B、甘草異黃酮B、芹糖甘草苷、甘草苷、甘草素、佛來心苷、芹糖異甘草苷、異甘草苷、新異甘草苷、異甘草素、甘草香豆素、甘草酚及甘草黃酮醇對照品適量,加甲醇溶解并稀釋成質量濃度分別為46.26、182.20、4.16、11.63、9.17、116.05、90.44、15.70、5.47、36.54、19.96、11.36、4.62、8.06、14.00、4.40 μg·mL-1的混合對照品溶液1。
精密稱取甘草查耳酮B、刺甘草查耳酮、甘草查耳酮A及光甘草酚對照品適量,加甲醇溶解并稀釋成質量濃度分別為21.48、2.87、43.90、18.66 μg·mL-1的混合對照品溶液2,即得。
2.1.3 供試品溶液的制備 取本品粉末(過三號篩)約0.5 g,精密稱定,置具塞錐形瓶中,精密加入70%甲醇25 mL,密塞,稱量,超聲(超聲功率:300 W,頻率:40 kHz)處理30 min,放冷,再稱量,用70%甲醇補足減失的質量,搖勻,濾過,取續濾液,即得供試品溶液1。
取本品粉末(過三號篩)約0.5 g,精密稱定,置具塞錐形瓶中,精密加入甲醇25 mL,密塞,稱量,超聲(超聲功率:300 W,頻率:40 kHz)處理45 min,放冷,再稱量,用甲醇補足減失的質量,搖勻,濾過,取續濾液,即得供試品溶液2。
2.1.4 方法學驗證 依據課題組已建立的HPLC 方法[9,16],經方法學驗證,20 個化合物的線性相關系數(r)均大于0.999 4,檢出限(LOD)和定量限(LOQ)分別為0.002 7~0.062 8 μg·mL-1和0.008 8~0.209 2 μg·mL-1,加樣回收率為91.19%~101.25%,穩定性、精密度和重復性RSD 分別為2.92%、1.00%和2.33%。方法學驗證結果符合《中華人民共和國藥典》2020 年版(四部)通則9101《藥品質量標準分析方法驗證指導原則》。
2.2 數據分析
聚類分析(HCA)和主成分分析(PCA)采用ChemPattern 2017 化學計量學軟件;t檢驗采用SPSS Statistics 23.0 軟件;正交偏最小二乘法-判別分析(OPLS-DA)采用SIMCA 13.0軟件。
3 結果
3.1 光果甘草與脹果甘草中20個化合物含量
采用外標法計算20批甘草樣品中20個化合物含量,見表2。分別統計光果甘草與脹果甘草中20 個化合物含量,結果見圖3。
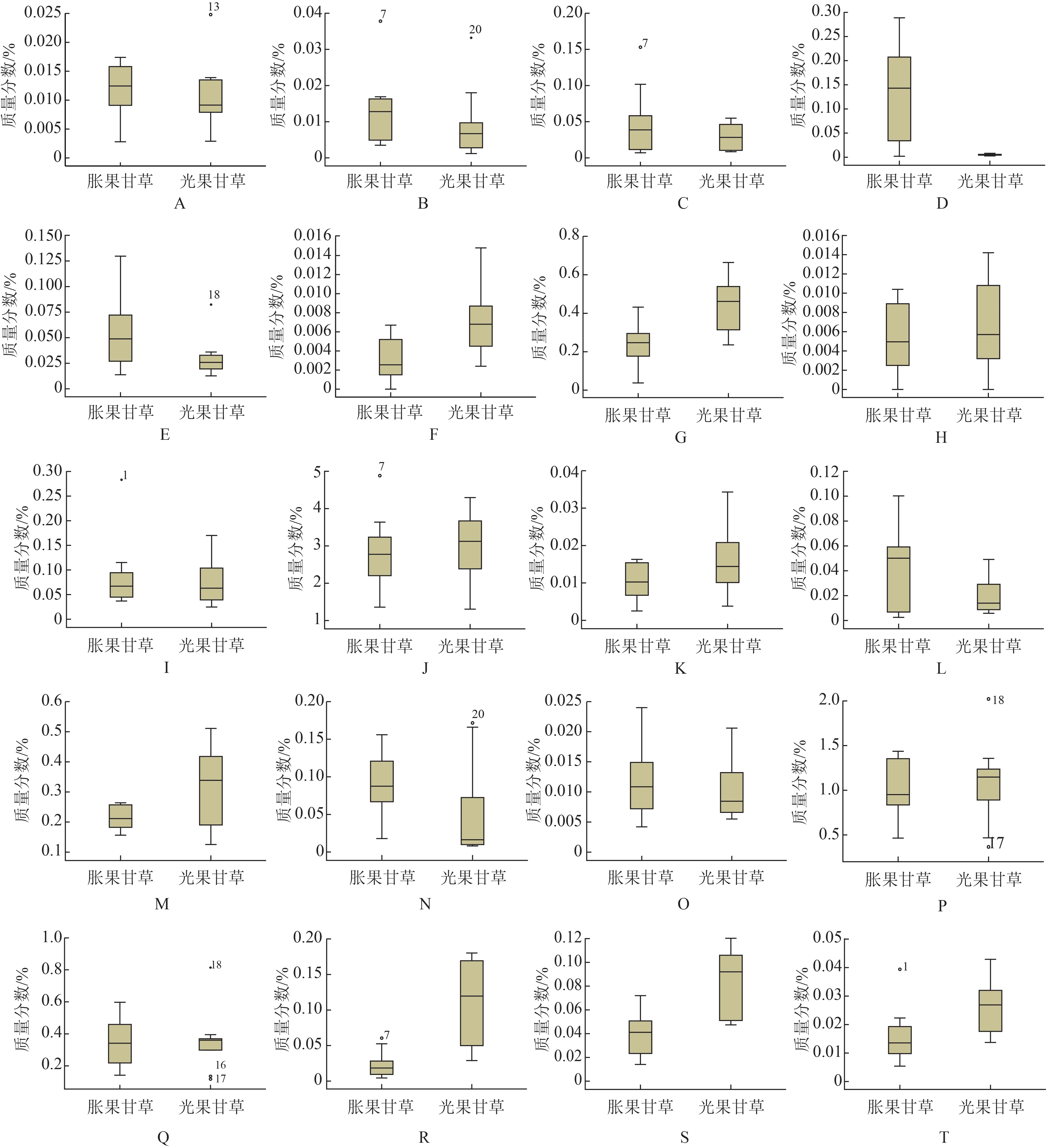
圖3 脹果甘草和光果甘草中20個化學成分含量分布(n=10)
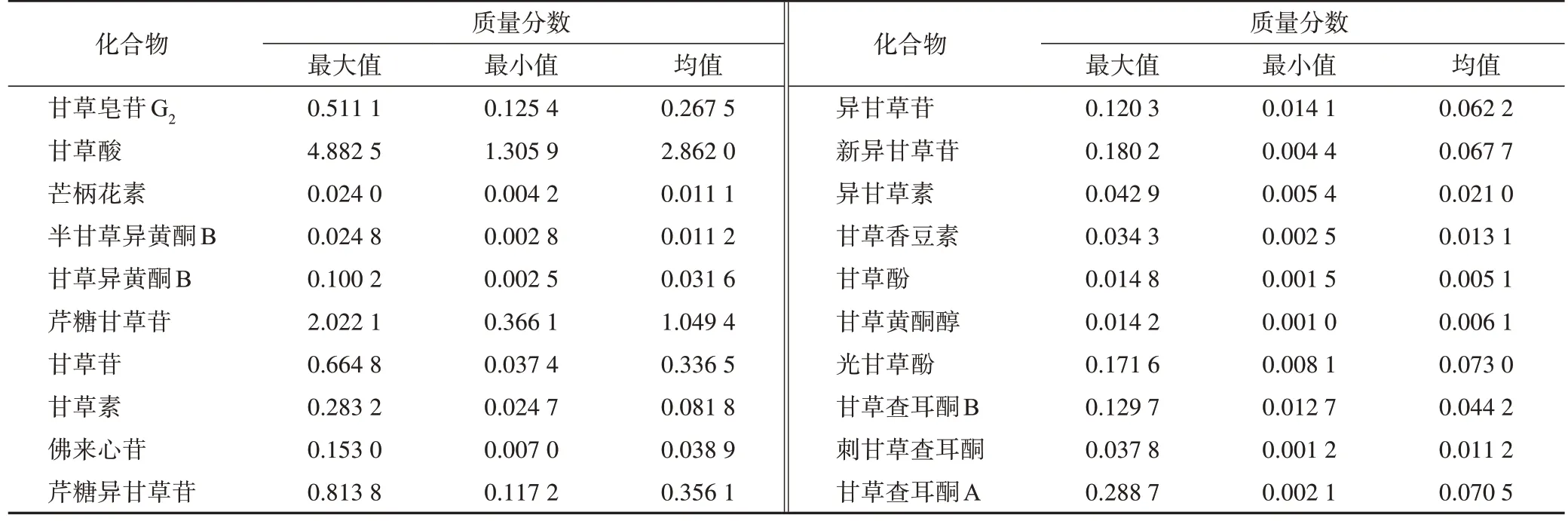
表2 甘草藥材中20個化學成分質量分數(n=20)%
3.2 HCA
利用Chem Pattern 軟件將光果甘草和脹果甘草20 個化學成分含量數據標準化后,以歐式距離為度量,采用遠鄰法進行聚類分析,見圖4。由圖4 可知,光果甘草和脹果甘草可以各自聚為一類。
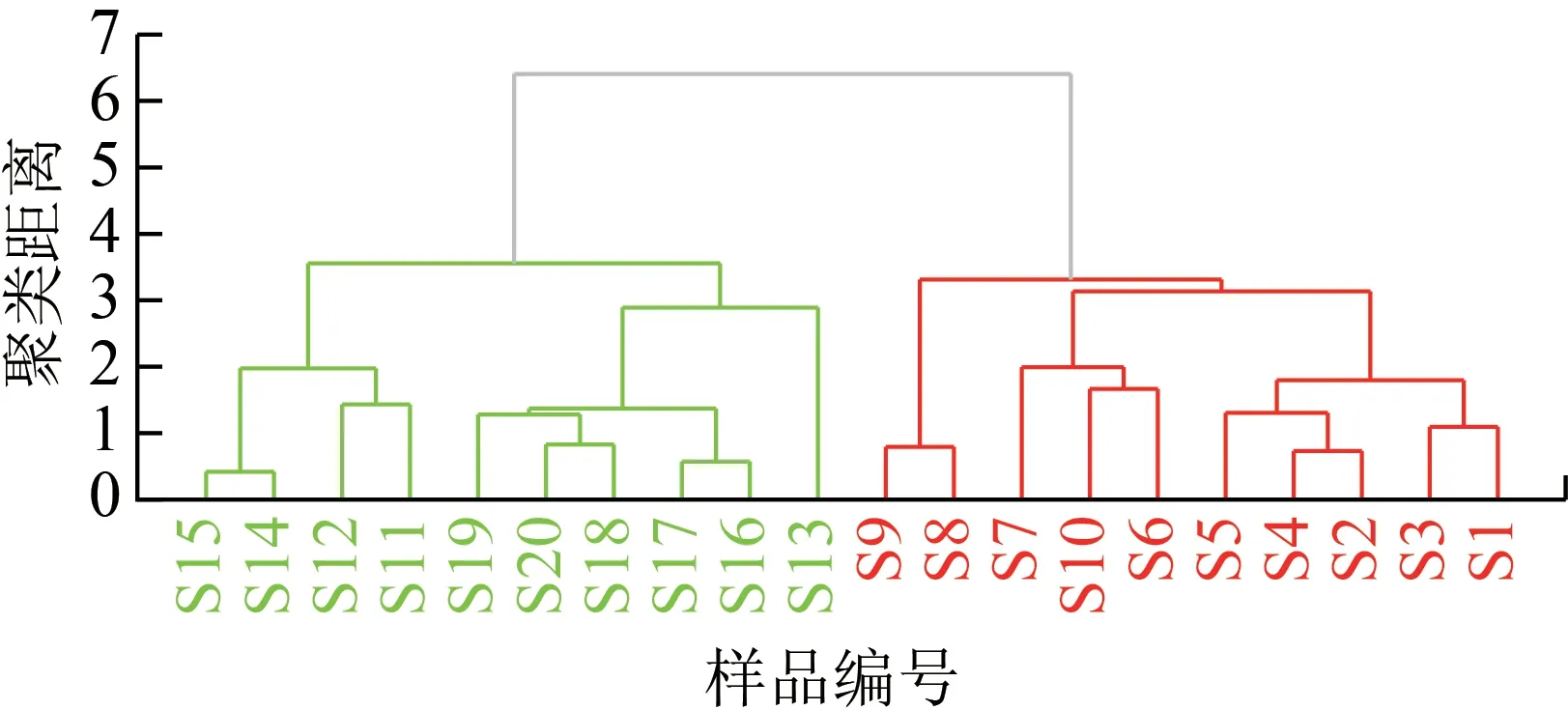
圖4 光果甘草和脹果甘草20個化學成分含量的HCA
3.3 PCA
以20個化學成分含量為變量,采用ChemPattern軟件進行主成分分析,見圖5。第一主成分(PC1)的方差貢獻率為73.30%,第二主成分(PC2)的方差貢獻率為18.59%,累積方差貢獻率為91.89%,說明其能較好地反映樣品間的差異。PCA 散點圖顯示,光果甘草和脹果甘草可以各自聚為一類,該結果與聚類分析結果相符。
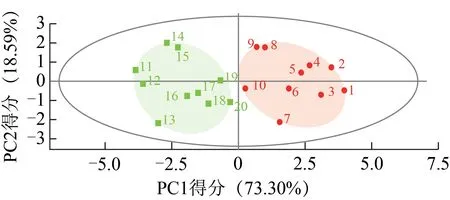
圖5 光果甘草和脹果甘草20個化學成分含量的PCA
3.4 OPLS-DA
采用SIMCA 13.0 軟件,以20 個化學成分含量為自變量,20批樣品為因變量,進行OPLS-DA。由圖6 可以看出,2 種基原的甘草藥材可以較好地區分,脹果甘草(S1~S10)集中分布于模型左側,光果甘草(S11~S20)集中分布于模型右側,OPLSDA 分類結果與HCA、PCA 結果一致[17-21]。采用交叉驗證法對模型進行驗證,模型對自變量擬合指數R2X=0.6,對因變量的擬合指標R2Y=0.915,模型預測指數Q2=0.802,三者均大于0.5,說明模型穩定可靠,可用于光果甘草和脹果甘草的區分。
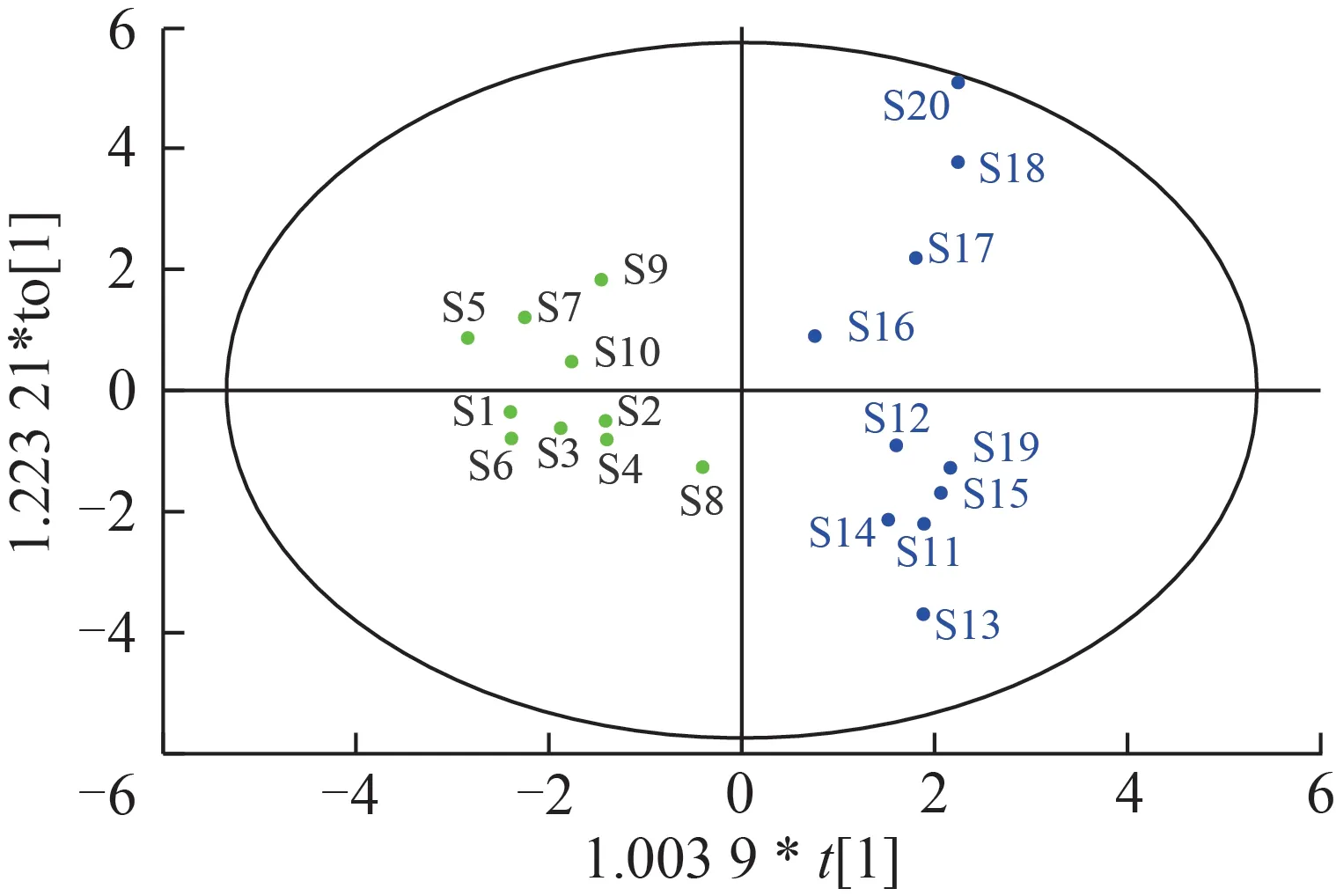
圖6 光果甘草和脹果甘草20個化學成分含量的OPLS-DA
為進一步找出影響2 種基原甘草藥材差異的主要變量,得到了OPLS-DA 模型的變量重要性投影(VIP)值,見表3。VIP值的大小代表了各指標成分對模型貢獻率的大小,值越大貢獻越大。以VIP值>1為界限進行篩選,新異甘草苷、異甘草苷、甘草查耳酮A、甘草苷、甘草酚、甘草皂苷G2、異甘草素和甘草查耳酮B 是影響光果甘草和脹果甘草差異的主要化學成分,對2 種基原甘草區分的影響程度為新異甘草苷>異甘草苷>甘草查耳酮A>甘草苷>甘草酚>甘草皂苷G2>異甘草素>甘草查耳酮B。
3.5 t檢驗
為驗證經OPLS-DA 找到的2 個基原甘草差異化學成分的準確性,采用SPSS Statistics 23.0軟件,按照基原類別將20 批樣品分為2 類,并以樣本類別為自變量,20個化學成分含量測定結果為因變量進行t檢驗,結果見表3[22]。新異甘草苷、異甘草苷、甘草查耳酮A、甘草苷、甘草酚、甘草皂苷G2、刺甘草查耳酮和異甘草素的含量在光果甘草和脹果甘草中差異有統計學意義(P<0.05)。
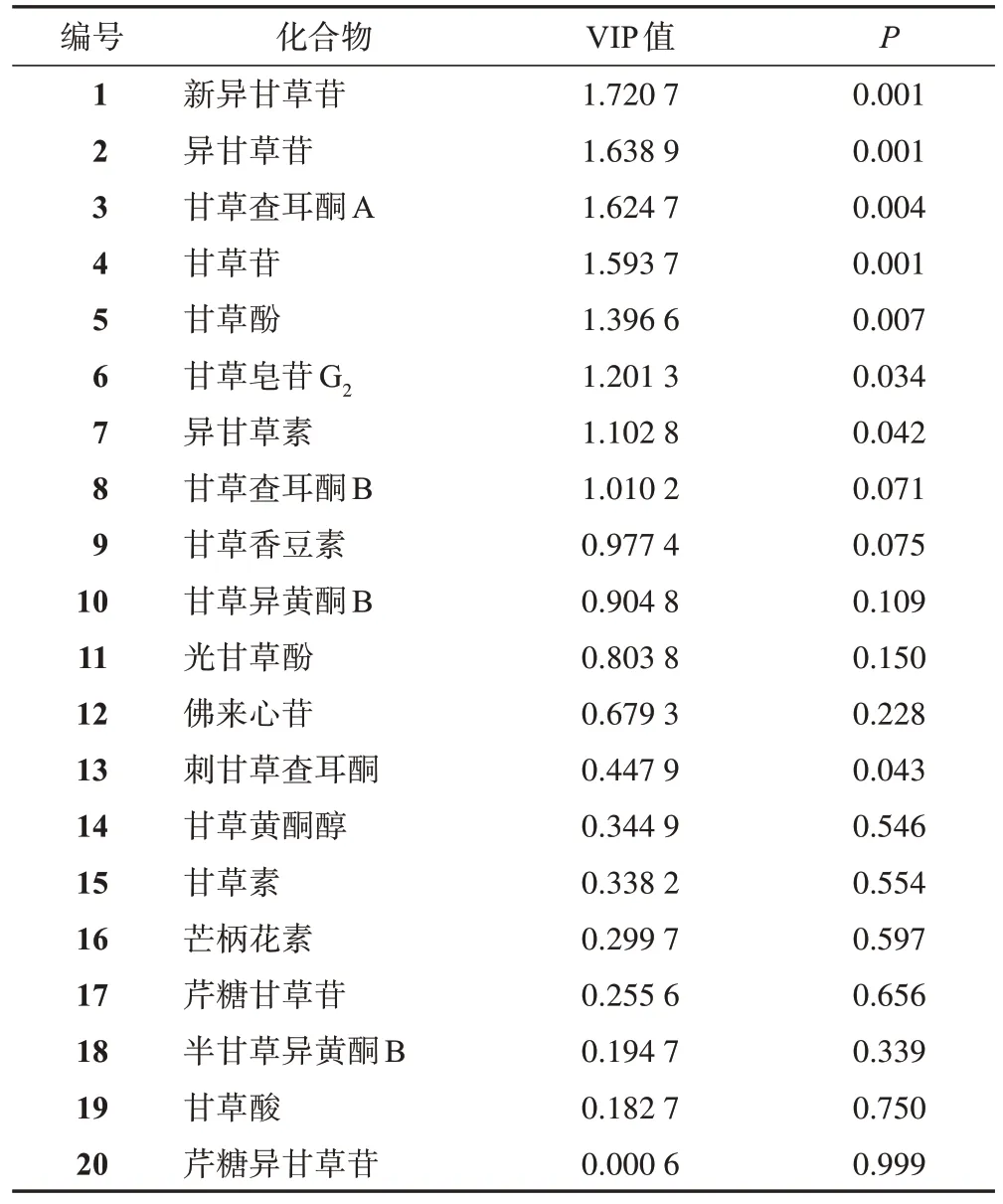
表3 光果甘草和脹果甘草20個化學成分含量OPLS-DA VIP值和t檢驗P值
對2 種統計方法的分析結果進行綜合分析,發現新異甘草苷、異甘草苷、甘草查耳酮A、甘草苷、甘草酚、甘草皂苷G2及異甘草素既對2 種基原甘草藥材OPLS-DA 模型貢獻大(VIP 值>1),又在t檢驗中差異有統計學意義(P<0.05);甘草查耳酮B雖在OPLS-DA 中對模型貢獻大(VIP 值>1),但在t檢驗中差異無統計學意義;刺甘草查耳酮雖在t檢驗中差異有統計學意義(P<0.05),但在OPLS-DA 中對模型貢獻較小(VIP值<1);甘草香豆素、甘草異黃酮B、光甘草酚、佛來心苷、甘草黃酮醇、甘草素、芒柄花素、芹糖甘草苷、半甘草異黃酮B、甘草酸和芹糖異甘草苷既對OPLS-DA模型貢獻較小(VIP值<1),又在t檢驗中差異無統計學意義。故將新異甘草苷、異甘草苷、甘草查耳酮A、甘草苷、甘草酚、甘草皂苷G2及異甘草素7 個化學成分作為區分2 個基原甘草藥材的差異化學成分。
4 討論
4.1 待測化學成分的選擇
甘草化學成分較復雜,以黃酮類和三萜皂苷類化學成分為主,另有香豆素類、二苯乙烯類等其他結構類型的化學成分,不同結構類型的化學成分藥理作用有所不同[23-24]。已有研究表明,光甘草定是光果甘草的特征化學成分,也是主要化學成分[25-26],但光果甘草中其他化學成分的研究報道較少。本研究選擇黃酮類、三萜皂苷類、香豆素類等不同結構類型的20 個化學成分作為研究對象,發現光果甘草和脹果甘草的差異化學成分包括4 個黃酮類化合物、1 個三萜皂苷類化合物、1 個香豆素類化合物和1 個查耳酮類化合物,說明不同結構類型的化合物含量在2個基原的甘草藥材中均存在不同程度的差異。
4.2 統計結果分析
本研究通過OPLS-DA 尋找光果甘草和脹果甘草的差異化學成分,發現甘草查耳酮B 在OPLS-DA 中VIP值>1,初步認為該化合物在2個基原甘草藥材中含量是有差異的,再采用t檢驗驗證發現,甘草查耳酮B 在2 個基原甘草藥材中的含量差異無統計學意義。采用同樣的統計方法,發現新異甘草苷等7 個化學成分既在OPLS-DA 中VIP>1,即對該模型貢獻大,又經t檢驗驗證發現其在2 個基原甘草藥材中的含量差異有統計學意義,因此最終確定該7 個化學成分是光果甘草和脹果甘草的差異化學成分。
4.3 差異化學成分分析
本研究找到的7 個差異化學成分在光果甘草和脹果甘草中均能檢出,屬于含量高低的區別。甘草查耳酮A 在脹果甘草中的含量高于光果甘草,甘草苷、異甘草苷和異甘草素在光果甘草中的含量高于脹果甘草,這與已有文獻報道一致[11,27]。本研究發現,新異甘草苷在光果甘草和脹果甘草中的含量差異有統計學意義,其在光果甘草中的含量高于脹果甘草。本課題組前期研究發現,新異甘草苷在甘草清熱解毒功效方面具有潛在生物活性[23],此外,在不同生長年限甘草中的含量也具有顯著差異[28],本課題組將對該化學成分開展深入研究,進一步考察其作為不同基原甘草藥材質量控制指標的可能性。
本課題組在前期研究中發現,甘草與脹果甘草和光果甘草存在明顯的化學成分差異。本研究進一步分析了脹果甘草和光果甘草化學成分的差異,發現甘草查耳酮A 等7 個化學成分在脹果甘草和光果甘草中差異有統計學意義。脹果甘草富含甘草查耳酮類成分,具有抗炎、抗腫瘤、抗氧化等作用,常被用于制備甘草提取物。光果甘草黃酮類成分具有抗炎及抑制酪氨酸酶的活性,是良好的黑色素抑制劑,常被用作美白類化妝品原料[29-31]。本研究結果為多基原甘草藥材質量評價及相關標準制定提供科學依據,同時也為多基原甘草藥材綜合開發利用提供參考。
猜你喜歡
英語世界(2023年10期)2023-11-17 09:19:16
汽車實用技術(2022年10期)2022-06-09 11:16:58
音樂探索(2022年2期)2022-05-30 21:01:37
收藏界(2019年3期)2019-10-10 03:16:40
小天使·一年級語數英綜合(2019年8期)2019-08-27 02:23:00
中國特種設備安全(2018年11期)2019-01-08 02:08:32
小學科學(學生版)(2018年7期)2018-08-13 09:33:04
中國非營利評論(2017年1期)2017-11-09 03:09:10
海外華文教育(2017年8期)2017-11-07 04:42:02
現代語文(2016年21期)2016-05-25 13:13:50
