固相微萃取-氣相色譜串聯兩級質譜法測定水中10種藻類代謝產物
2022-02-10 05:34:40景二丹許小燕顧青清余沛芝張琳琳
凈水技術 2022年2期
景二丹,許小燕,顧青清,余沛芝,張琳琳,李 霞
(蘇州工業園區清源華衍水務有限公司,江蘇蘇州 215000)
隨著水體富營養化問題越來越嚴重,藻類水華事件越來越頻繁。給水系統是一項重要的城市公共服務設施,藻類滋生引起的水質污染問題對城市居民的健康危害極大[1]。其中,嗅味物質作為藻類主要的次代謝產物,越來越多地在飲用水中被檢測出來。嗅和味作為評價飲用水的重要指標,這類物質所產生的異味嚴重影響了人們對水質的感覺和評價,近年來已逐漸成為水質改善研究的熱點[2]。自然水體中常見的嗅味物質有萜類、脂肪類以及代謝產物[3]。主要異味有土霉味、芳香味、魚腥味、草木味、沼氣味、薄荷味、腐敗味。2-甲基異莰醇、土臭素、三氯苯甲醚、甲氧基吡嗪等物質產生土霉味;β-環檸檬醛產生草木味;β-紫羅蘭酮產生紫羅蘭味;異氟爾酮產生薄荷味[4-6]。
隨著人們對水體中嗅味物質越來越關注,供水行業對水體中嗅味物質的檢測越來越重視。《生活飲用水衛生標準》(GB 5749—2006)規定2-甲基異莰醇、土臭素的檢出限為10 ng/L。目前,世界各國對于其他幾種物質的限值尚無統一規定,含量一般控制在1~10 ng/L[7]。測定嗅味物質的方法有頂空固相微萃取-氣相色譜法[2]、吹掃捕集-氣相色譜質譜法[8-9]、固相萃取-氣相色譜質譜法[10]、固相微萃取-氣相色譜質譜法[11-12]。其中,吹掃捕集-氣相色譜-質譜聯用法和頂空固相微萃取-氣相色譜法的檢出限整體較高;固相萃取的前處理使用二氯甲烷做萃取劑,毒性較大、污染環境且耗費時間較長。通過建立一種固相微萃取-氣相色譜串聯兩級質譜儀,聯用測定水中10種藻類代謝產物的含量,此方法操作簡單、檢出限低、耗費時間短、靈敏度高、無副作用,可以滿足實際水樣檢測的相關研究的要求。
1 試驗部分
1.1 儀器與試劑
Agilent 7890B-7000C型氣相色譜/質譜/質譜儀(安捷倫);德國Gerstel多功能樣品前處理平臺MPS XT。
固相微萃取纖維:50/30 μm DVB/CAR/PDMS,Supelco;65 μm PDMS/DVB,Supelco;Milli-Q純水器。
標準物質:2-甲基異莰醇、土臭素、2,4,6-三氯苯甲醚、2,3,4-三氯苯甲醚、2,3,6-三氯苯甲醚、2-異丙基-3-甲氧基吡嗪、2-異丁基-3-甲氧基吡嗪均為色譜純(上海安譜實驗科技股份有限公司生產,100 mg/L);β-環檸檬醛(Sigma,98%,1 mL,0.947 g/cm3)、β-紫羅蘭酮(Sigma,98%,1 mL,0.945 g/cm3);異氟爾酮(德國Dr,99.3%,100 mg,0.926 g/cm3)。甲醇為色譜純;氯化鈉為分析純。
1.2 儀器條件
1.2.1 固相微萃取條件
萃取溫度為65 ℃;萃取時間為25 min;萃取深度為25 mm;解析時間為3 min;加熱器轉速為250 r/min。
1.2.2 色譜-質譜條件
HP-5MS毛細管色譜柱(30 m×0.25 mm,0.25 μm,美國Agilent公司);載氣為高純He(純度為99.99%),不分流進樣;進樣口溫度為250 ℃;隔墊吹掃流量為3.0 mL/min;柱箱溫度:起始溫度為40 ℃,保持3 min,然后以10 ℃/min升溫到180 ℃,保持1 min,最后以30 ℃/min升溫到240 ℃,保持4 min。
色譜柱流量:起始流速為1.3 mL/min;運行流速為1.2 mL/min。進樣口壓力為0.688 Pa,載氣為He,載氣流量為1.3 mL/min,出樣檢測器為MSD,柱溫箱溫度的初始值為40 ℃;平均線速度為41.345 cm/s;滯留時間為1.209 3 min。
1.2.3 質譜條件
離子源溫度為230 ℃;四級桿溫度為150 ℃;質譜檢測器溫度為280 ℃。
QQQ 碰撞池前端(EPC):載氣He(淬滅氣體)流量為2.25 mL/min;載氣N2(碰撞氣體)流量為1.5 mL/min。采用選擇離子掃描(SIM)模式定量,質譜參數如表1所示。
1.3 試驗方法
標準使用溶液(10.00 mg/L)的制備:精確稱取0.100 0 mg的β-紫羅蘭酮,分別移取106、110 μL的β-環檸檬醛和異氟爾酮,用甲醇定容于10 mL容量瓶中。
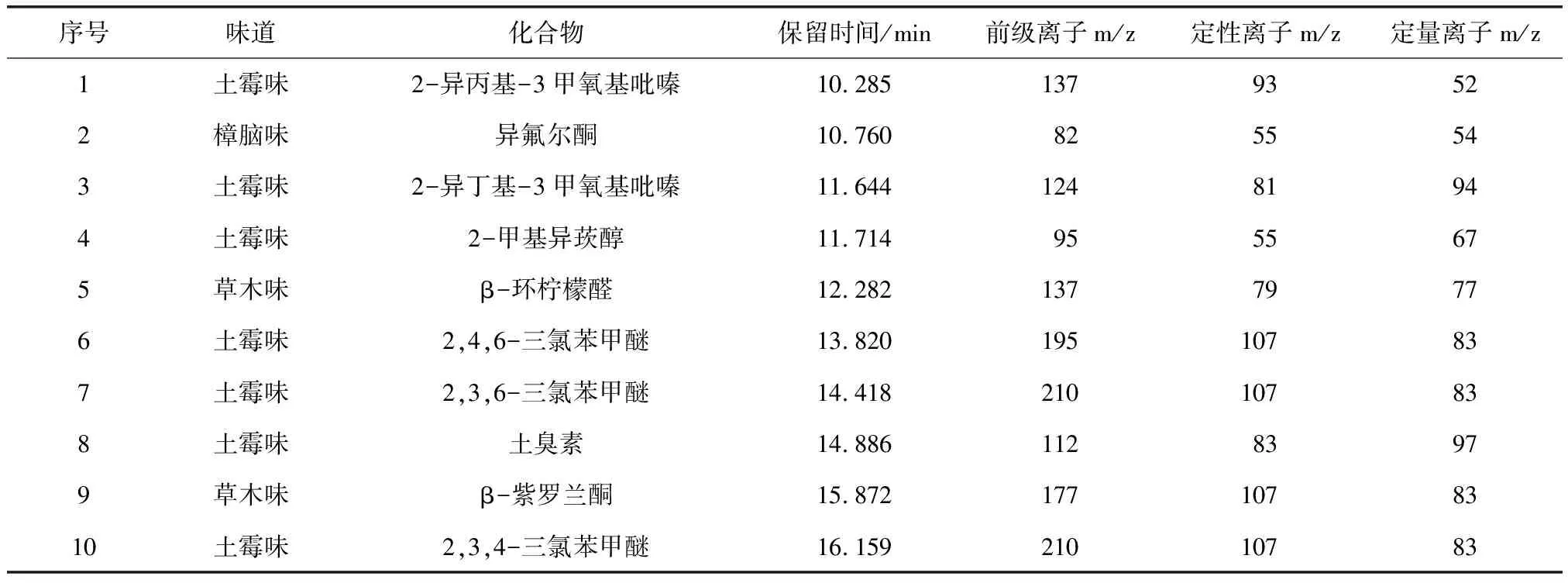
表1 10種物質的保留時間和特征離子Tab.1 Retention Time and Characteristic Ions of 10 Substances
移取100 mg/L的2-甲基異莰醇、土臭素、2,4,6-三氯苯甲醚、2,3,4-三氯苯甲醚、2,3,6-三氯苯甲醚、2-異丙基-3甲氧基吡嗪、2-異丁基-3甲氧基吡嗪100 μL,用甲醇定容于10 mL容量瓶中。
稱取1.7 g氯化鈉加入20 mL頂空瓶中,然后取水樣或標液10 mL,迅速蓋上瓶蓋,搖勻,放置于樣品盤上待測。置樣品瓶于多功能樣品前處理平臺MPS2樣品盤上進行固相微萃取,萃取結束后,插入氣相色譜-質譜儀進樣口解析進樣(3 min)。
2 結果與討論
2.1 萃取條件的選擇
2.1.1 萃取纖維的選擇
試驗對比了50/30 μm DVB/CAR/PDMS和65 μm PDMS/DVB這兩種萃取纖維對目標物的萃取效率,結果如圖1所示。50/30 μm DVB/CAR/PDMS對10種目標物有較好的萃取效果,65 μm PDMS/DVB對β-紫羅蘭酮的富集效果差。因此,選擇50/30 μm DVB/CAR/PDMS作為萃取纖維。
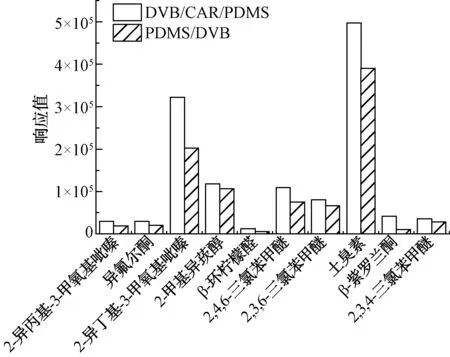
圖1 不同萃取纖維的萃取效果Fig.1 Effect of Different Extraction Fibers
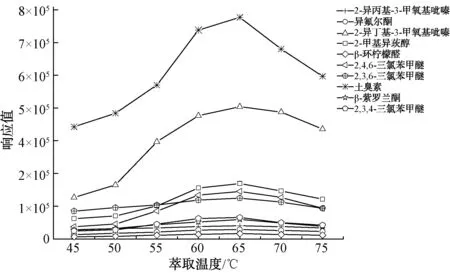
圖2 不同萃取溫度的萃取效果Fig.2 Effect of Different Extraction Temperatures
2.1.2 萃取溫度的選擇
試驗考察了萃取溫度對試樣達到氣液平衡的影響,從45 ℃起以5 ℃遞增至75 ℃,結果如圖2所示。待測物質氣相組分在45~65 ℃顯著增加,65 ℃以后有下降趨勢,因此,選擇65 ℃作為最佳平衡溫度。
2.1.3 萃取時間的選擇
平衡時間也是影響待測物質氣液分配比的一個重要因素。配制20.00 ng/L混合標準溶液,在其他條件不變的情況下,分別選取平衡時間為10、15、20、25、30、35、40、50 min,結果如圖3所示。當萃取時間在10~25 min時,樣品響應值顯著增加,25 min以后,其目標物的響應值處于水平。因此,選擇25 min作為最佳萃取時間。
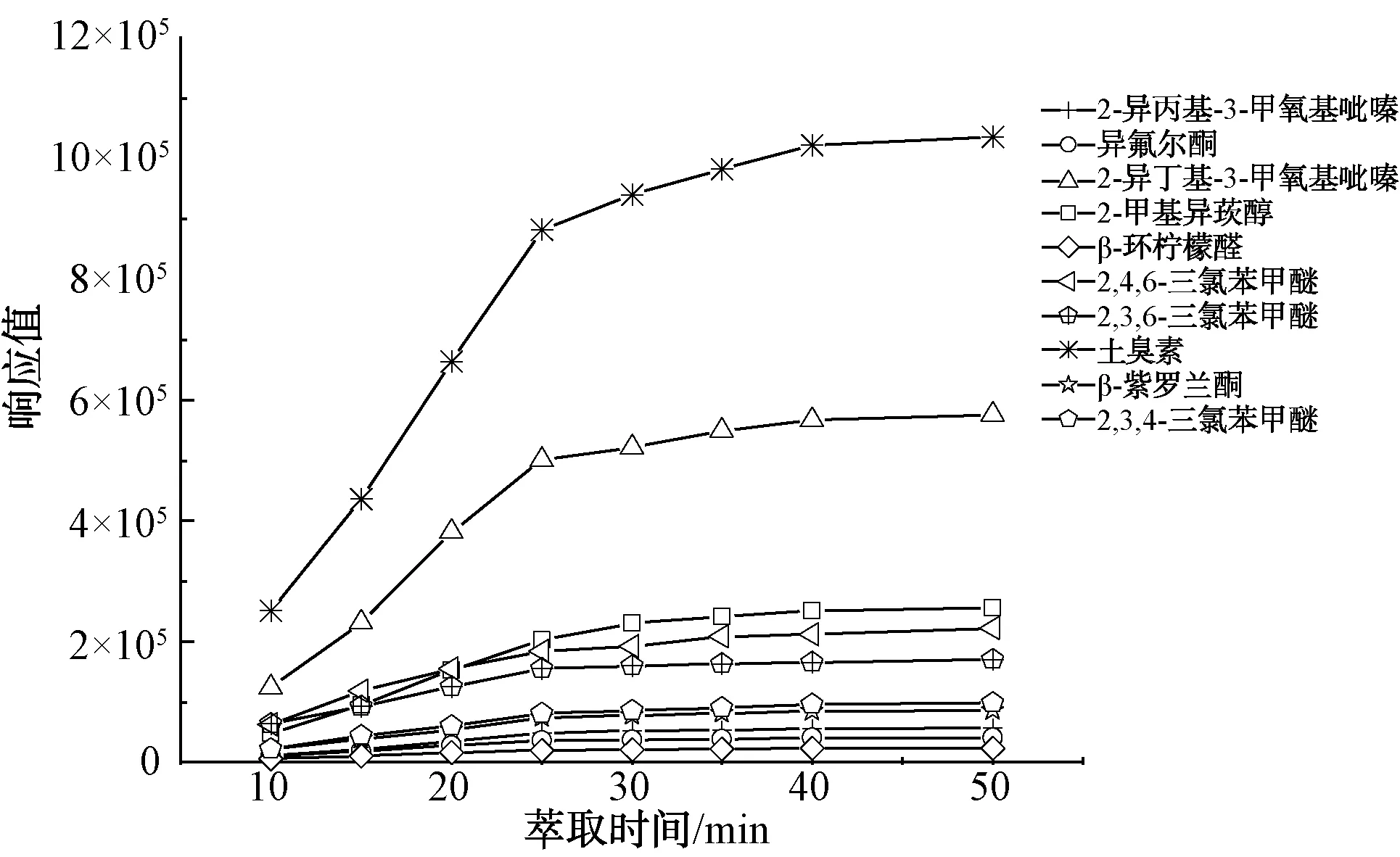
圖3 不同萃取時間的萃取效果Fig.3 Effect of Different Extraction Times
2.1.4 萃取劑量的選擇
如圖4所示,水樣中加入氯化鈉可以促進目標物揮發,有利于提高萃取率,隨著氯化鈉含量的增加,萃取率呈增長趨勢。在氯化鈉含量為1.7 g時,萃取率增加較大,隨著繼續增加氯化鈉,增加幅度較平緩。因此,氯化鈉為1.7 g時是較理想的萃取劑量。
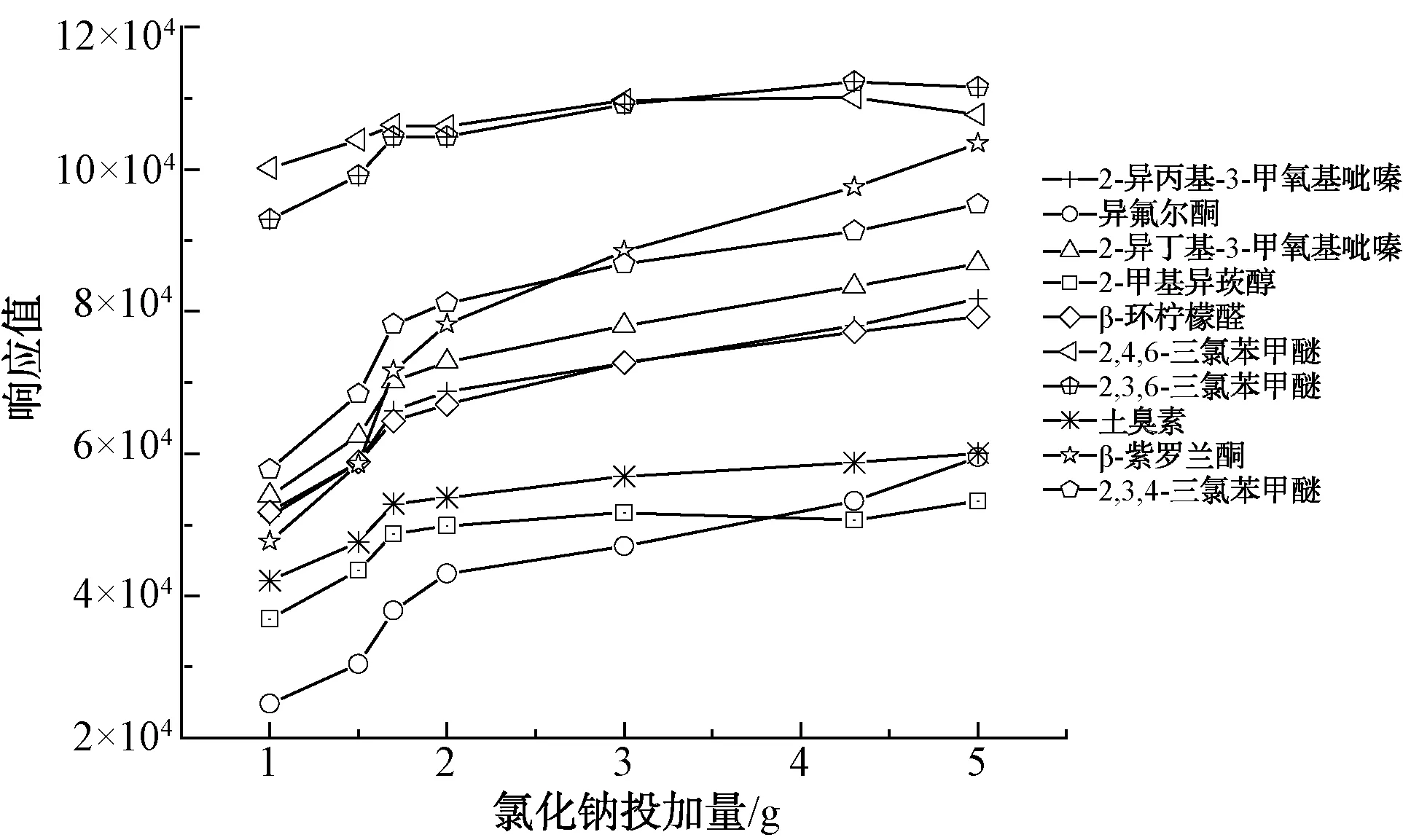
圖4 不同萃取劑量的萃取效果Fig.4 Effect of Different Extraction Doses
2.2 色譜柱的選擇
色譜柱關系到物質的分離效果,試驗分別選擇HP-5MS(填料為5%苯基-甲基聚硅氧烷)、HP-1MS(填料為100%甲基聚硅氧烷)兩種試驗用色譜柱,參照試驗方法分析質量濃度為20.00 ng/L的混合標準溶液。如圖5和圖6所示:采用HP-1MS色譜柱時,目標化合物2-異丁基-3甲氧基吡嗪和2-甲基異莰醇分離不完全,峰有部分重疊,不利于定量分析;在HP-5MS色譜柱上得到的分離效果和峰形較好。因此,最終選擇HP-5MS色譜柱。
2.3 標準曲線與方法檢出限
在選定的條件下,按試驗方法對10種混合標準溶液進行進樣分析,在5.00~100.00 ng/L線性回歸方程良好,如表2所示。考察方法檢出限,信噪比為2∶1時,檢出質量濃度為0.14~1.00 ng/L。

表2 10種藻類代謝產物的回歸方程、相關系數和檢出限Tab.2 Regression Equations, Correlation Coefficients and Detection Limits of 10 Algae Metabolites
2.4 回收率與精密度
在10.00 mL純水中進行加標回收試驗,標準加入量分別為20.00、50.00 、80.00 ng/L,按試驗方法分別平行測定7次,結果如表3所示。10種混合溶液的回收率均在90%~110%,異氟爾酮易揮發,在預處理以及加標時需現用現配。精密度RSD在9%以下,滿足檢測要求。
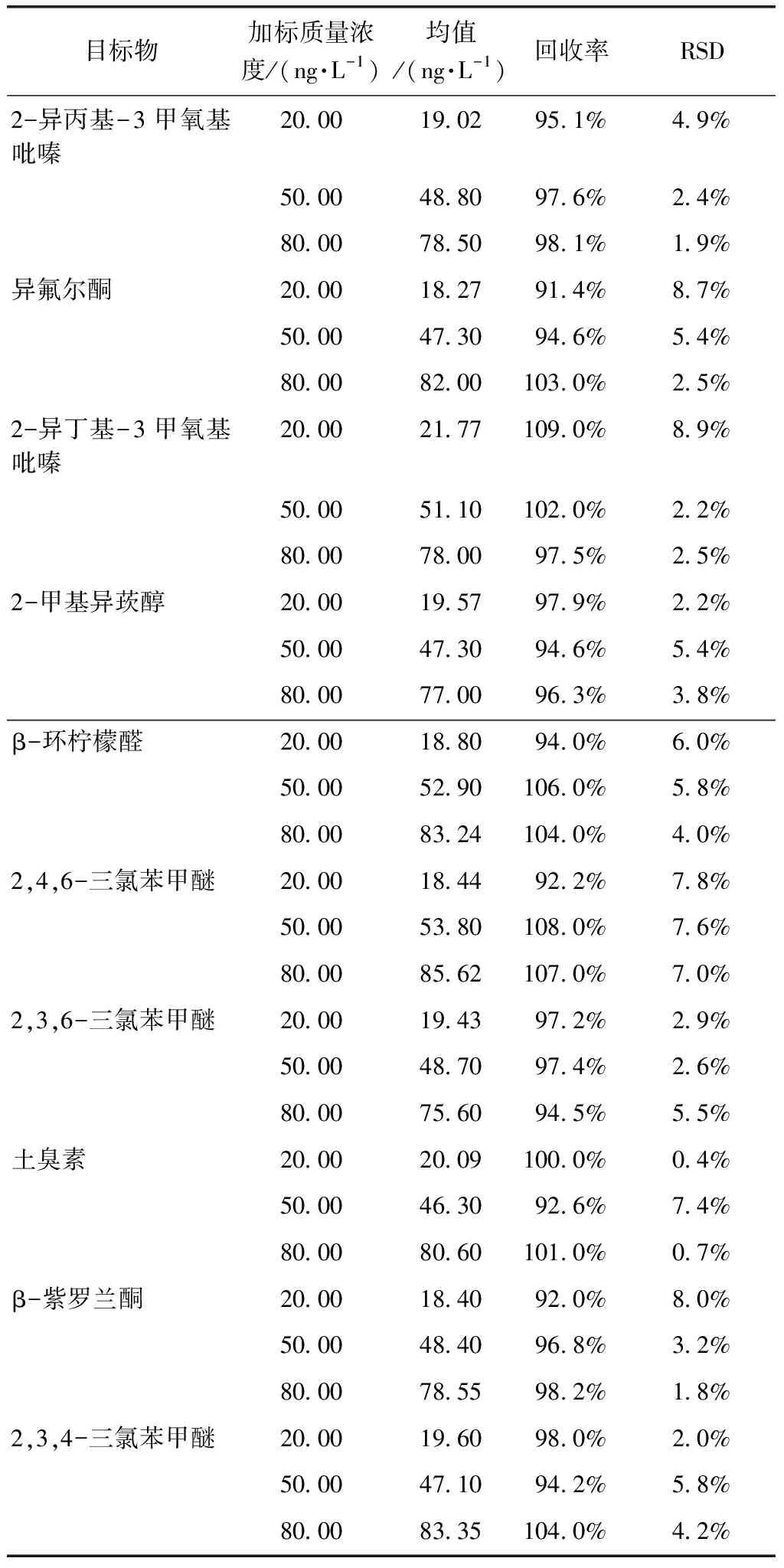
表3 純水回收率和精密度試驗結果Tab.3 Experimental Results of Pure Water Recovery and Precision
分別取10 mL地表水和出廠水進行加標測回收率,標準物質質量濃度為20.00 ng/L,按試驗方法分別平行測定7次,結果如表4所示。由于余氯對2-甲基異莰醇、β-環檸檬醛、β-紫羅蘭酮有干擾,出廠水需加50 μL硫代硫酸鈉(濃度為1.5%)于100 mL水樣中消氯再進行檢測。地表水的平均回收率為84.9%~100.4%,出廠水的平均回收率為81.95%~101.8%,滿足地表水和飲用水的檢測要求。
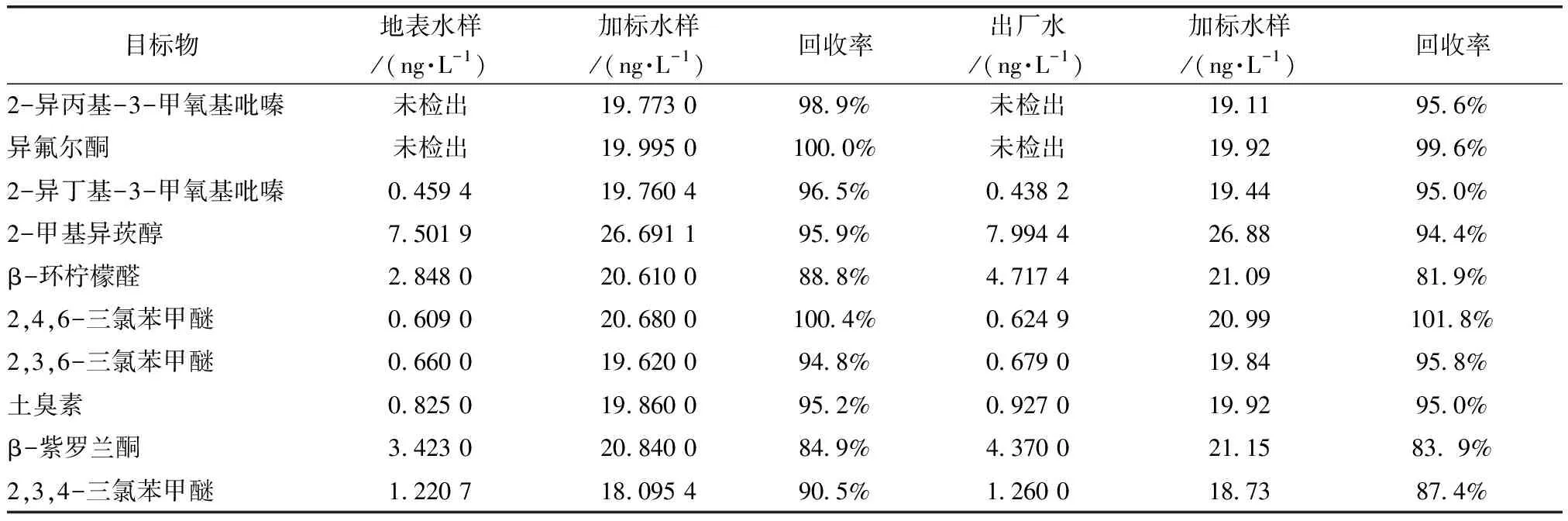
表4 地表水和出廠水回收率試驗結果Tab.4 Results of Surface Water and Finished Water Recovery Experiments
2.5 實際水樣分析
選取蘇州地區陽澄湖原水和X水廠的出水,按照試驗方法進行分析檢測,除2,4,6-三氯苯甲醚、2,3,6-三氯苯甲醚、2,3,4-三氯苯甲醚這3種物質未檢出外,其余7種物質檢測結果如表5所示。可以看出,10種藻類代謝產物的濃度都是極低的,用固相微萃取-氣相色譜串聯兩級質譜法對于低含量的嗅味物質也能很好地檢測,有利于水廠更好地監測水質。
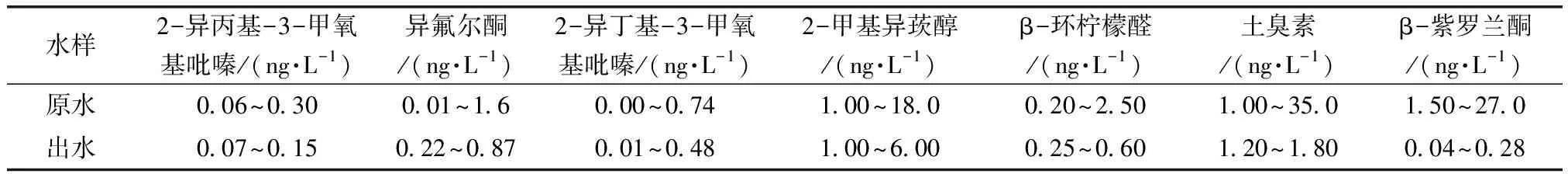
表5 實際水樣結果Tab.5 Results of Actual Water Samples
2.6 與文獻方法的比較
將所建立的方法與文獻報道的頂空/固相微萃取氣相色譜串聯質譜法、吹掃捕集氣相色譜串聯質譜法和固相萃取-氣相色譜串聯質譜法等方法進行對比。吹掃捕集氣相色譜串聯質譜法操作復雜,干擾多,回收率低。固相萃取-氣相色譜串聯質譜法操作步驟繁多(包括活化、上樣、洗滌、洗脫和濃縮等),所需時間較長,且在濃縮過程中易造成分析物的損失,洗脫劑有毒有害。本方法同時測定10種藻類代謝產物,具有線性范圍寬、檢出限低、無毒無害等優點。
由表6可知,在頂空/固相微萃取氣相色譜串聯質譜法和吹掃捕集氣相色譜串聯質譜法的檢測中,均沒有檢測到異氟爾酮,但根據實際水樣的檢測結果,此物質的濃度相對較高。固相萃取-氣相色譜串聯質譜法雖然可以檢測到異氟爾酮,但此法的線性范圍和檢出限均在微克級。本文的靈敏度較固相萃取-氣相色譜串聯質譜法提高1 000倍,更適合地表水和出廠水的檢測。
3 結論
本文建立了固相微萃取-氣相色譜串聯兩級質譜法測定水中10種藻類代謝產物的方法,在萃取時間為25 min、萃取溫度為65 ℃、氯化鈉含量為1.7 g的條件下,采用50/30 μm DVB/CAR/PDMS萃取頭進行預處理,HP-5MS色譜柱進行分離,質譜檢測器進行測定,可以使檢測結果達到最優效果。方法的質量濃度均達到5.00 ~100.00 ng/L,工作曲線回歸方程的相關系數R2>0.995;方法檢出限(2S/N)為0.14~1.00 ng/L。在加標量為20.00、50.00、80.00 ng/L時,平均回收率為92.0%~109.0%,精密度RSD為0.40%~8.90%,實際水樣的平均回收率為83.9%~101.8%,滿足地表水和飲用水的檢測要求。
該方法操作過程簡單、干擾少、預處理簡單、無污染、無毒性、檢測效率高、檢出限低(ng級),靈敏度較固相萃取-氣相色譜-質譜法提高1 000倍。該方法適合低含量的藻類代謝產物的測定,準確度高,適用于水中10種藻類代謝產物的分析研究。
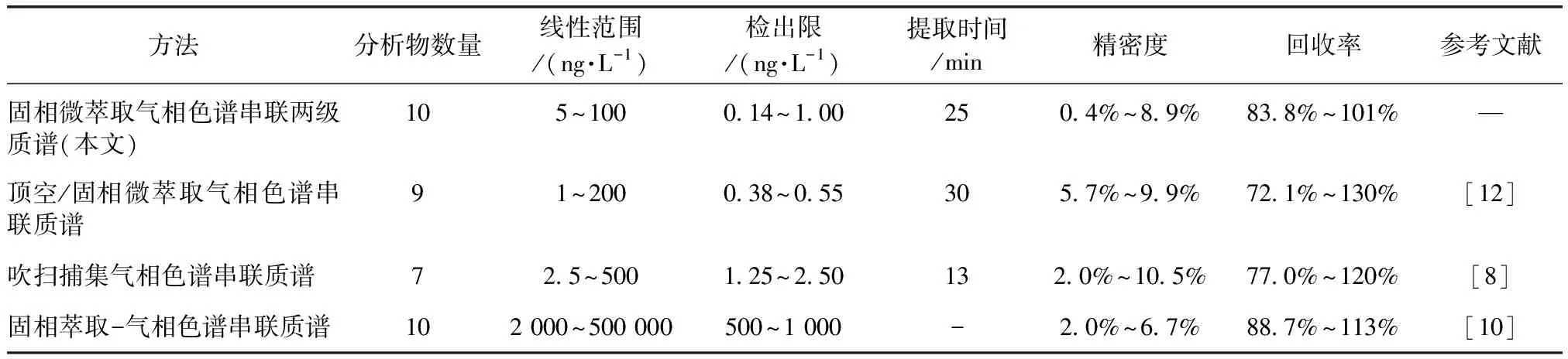
表6 本方法與其他方法的比較Tab.6 Comparison of the Method with Others
猜你喜歡
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
兒童故事畫報(2019年5期)2019-05-26 14:26:14
海峽科技與產業(2016年3期)2016-05-17 04:32:12
Coco薇(2016年2期)2016-03-22 02:42:52
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年7期)2015-08-11 15:03:12
