睡眠調控與睡眠調節藥物研究進展
2022-02-15 08:20:04崔素穎張永鶴
中國藥理學與毒理學雜志 2022年11期
關鍵詞:系統
崔素穎,秦 宇,張永鶴
(北京大學醫學部學基礎醫學院藥理學系,北京 100191)
從古希臘亞里士多德時代到20世紀初,大多數哲學家和醫學家都認為睡眠只是外界感覺輸入減少和大腦活動度降低的結果,即認為睡眠是覺醒的副產物,屬于覺醒之后的被動過程。然而,隨著研究人員在大腦中發現越來越多的“促睡眠神經元”和“促覺醒神經元”,這些神經元構成精密的睡眠-覺醒神經調控網絡,并以此為基礎調整機體新陳代謝、情緒重塑和認知體系時,人們逐漸意識到“睡眠被動學說”的不足,并開始接納“睡眠主動學說”,即睡眠絕不僅僅是覺醒的附屬物,而是維持人類生命不可或缺的、為了生存而演化的無意識的行為范式[1]。
隨著人們生活節奏加快和工作壓力激增,睡眠疾病已成為臨床中最常見疾病,嚴重影響患者的身心健康,以及由此導致的意外傷害、工作效率低下也給社會帶來了負面影響。但比較令人鼓舞的是,光遺傳學實驗技術、基因修飾動物模型以及人腦功能性成像技術的普及和應用,助推了睡眠醫學研究的高速發展,使我們在時間和空間維度,更準確、更清晰、更精細地捕捉到“促睡眠神經元”和“促覺醒神經元”的生物學特性以及它們的運行模式,這也促進了睡眠疾病治療藥物的研發。近10年來,基于新機制、作用于新靶點的多種睡眠調節藥物也陸續進入臨床,用于治療睡眠疾病。
本文基于近年來取得的睡眠醫學研究進展,對睡眠-覺醒神經調控網絡以及擾動該網絡的晝夜節律機制、內穩態機制及睡眠調節藥物的研究進展加以綜述,旨在進一步了解“促睡眠神經元”和“促覺醒神經元”的生物學特性,以及這些神經元調控睡眠-覺醒行為的神經機制,并把握睡眠疾病治療藥物的研究動態,為睡眠疾病的病因學和治療學研究提供可借鑒的資料。
1 睡眠-覺醒調控理論
1.1 睡眠-覺醒相位調節雙過程模型
1982年,瑞士科學家 Alexander Borbély關于睡眠調控過程提出了“雙過程理論模型”(twoprocess model)[2]。該理論模型認為,睡眠是晝夜節律系統(circadian rhythm,process C)與機體內穩態系統(sleep-wake homeostasis,process S)相互作用整合進而調控睡眠需求(sleep need)的結果。晝夜節律對睡眠-覺醒行為的調節反映外界環境因素對覺醒的需求,睡眠穩態調節反映機體內部因素對睡眠的需求。主要表現為睡眠和覺醒行為隨晝夜變化而產生節律性振蕩,而睡眠的時長和深度與先前的覺醒時間相關[3]。
1.1.1 睡眠-覺醒行為的晝夜節律調節
機體可根據外界環境靈活調節晝夜節律,如倒班、倒時差,這是機體適應外部環境的表現。Scammell等[4]發現,機體的睡眠-覺醒調節系統和生物鐘系統之間存在廣泛的、間接的神經纖維投射,并揭示了在外部環境誘因下,機體的睡眠-覺醒行為發生晝夜節律倒換的神經生物學機制。視交叉上核(suprachiasmatic nucleus,SCN)神經元被認為是中央生物鐘核心腦區,這些神經元接受來自視網膜光敏細胞的興奮性傳入,其電活動呈現晝夜節律震蕩。但是SCN的神經元和“促覺醒神經元”、“促睡眠神經元”之間的神經纖維聯系較少,提示由SCN主導的生物鐘信息可能并不直接傳遞給睡眠-覺醒調控系統[5]。研究人員在外部環境變化誘導的睡眠-覺醒晝夜節律倒換的實驗動物中發現,SCN神經元的電活動始終與外界光刺激有關,而“促覺醒神經元”和“促睡眠神經元”的活動始終與動物的覺醒和睡眠狀態有關[6]。這進一步提示SCN與睡眠-覺醒調控系統之間的聯系并不是單突觸連接。由此推論,在倒班、倒時差等睡眠-覺醒晝夜節律倒換過程中,機體需要一個“轉換系統”重置晝夜節律對睡眠-覺醒行為的調節。后續的神經解剖學研究提示,下丘腦背內側核(dorsal medial hypothalamus,DMH)的神經元可能是這個“轉換系統”的一個中轉站,因為DMH神經元接受來自SCN直接通路的抑制性調控和來自SCN間接通路的興奮性調控。此外,DMH神經元還分別通過興奮性輸出和抑制性輸出,對“促覺醒神經元”和“促睡眠神經元”進行差異化調控[7]。DMH神經元的損傷或功能缺失,可以引起實驗動物睡眠-覺醒等多種生物功能的節律紊亂,同時伴有“促覺醒神經元”和“促睡眠神經元”的“失靈”[8]。
基于上述發現,研究人員提出睡眠-覺醒晝夜節律倒換的神經調控理論(圖1)。該理論認為,對于夜行性動物(如鼠類),白天外界光照信息激活SCN神經元,SCN通過直接通路的抑制性纖維投射,抑制DMH神經元,此時覺醒發生系統功能下調,睡眠發生系統功能上調,機體處于睡眠狀態;夜間,因SCN失去光信號的刺激作用,傳遞與日間相反的信息,機體進入覺醒狀態。然而,對于晝行性動物,SCN對DMH的輸出主要是通過途經室旁下核或SCN內部的間接通路完成,此時DMH最終接受興奮性傳入,動物表現出相反的睡眠-覺醒晝夜節律[9-10]。正是由于SCN的生物鐘系統對睡眠-覺醒系統存在多種調控路徑,才保證了機體根據外界環境的變化及時調整睡眠-覺醒節律。但該理論還存在尚未解釋清楚的問題。例如,機體如何切換SCN至DMH的直接通路和間接通路?分別投射至“促睡眠神經元”和“促睡眠神經元”的DMH神經元的生物學特點是什么?以及光信號對睡眠-覺醒晝夜節律的調節是否還有其他調節通路呢?針對后面的問題,復旦大學黃志力課題組發現,皮質下視覺反射中樞上丘的γ-氨基丁酸(γ-aminobutyric acid,GABA)能神經元接受來自視網膜神經節細胞的信號,并傳遞給腹側背蓋區多巴胺能神經元,這條通路在小鼠急性黑暗暴露誘發的促覺醒效應中發揮了關鍵性作用[11],這有望為光照異常導致的晝夜節律紊亂性睡眠疾病的發病機制提供新見解。
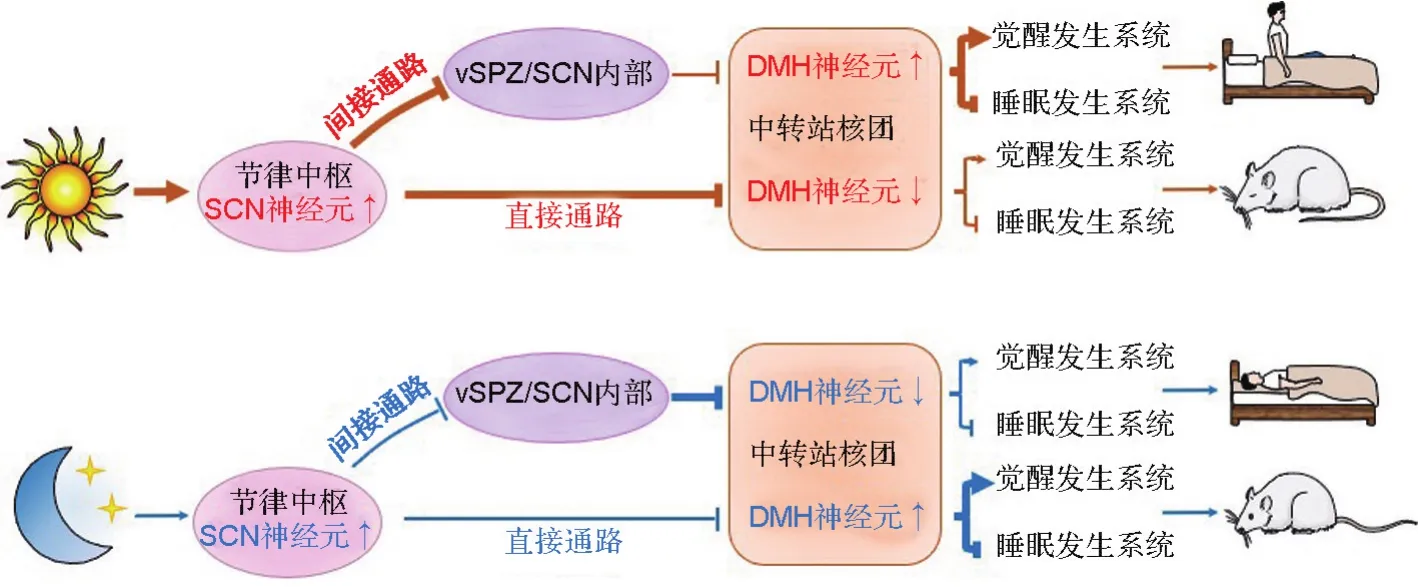
圖1 睡眠-覺醒晝夜倒換神經調控示意圖.SCN:視交叉上核;vSPZ:腹側腦室下區;DMH:下丘腦背內側核;:興奮增強;:興奮減弱;:抑制增強;:抑制減弱.
1.1.2 睡眠-覺醒行為的穩態調節
睡眠穩態過程是指隨覺醒時間延長,睡眠壓力逐漸增加成為“睡眠負荷”,而為了減輕“睡眠負荷”,機體會主動進入睡眠狀態的調節過程。因覺醒時間延長(如睡眠剝奪)引起的睡眠負荷積聚,需通過后續睡眠時間的延長或睡眠深度的提高(慢波活動的強化)得以消除。“睡眠負荷”可能是一種化學物質,如腺苷、褪黑素(melatonin,MT)或前列腺素D2,也可能是一種生物學效應,如神經突觸穩態、DNA損傷和蛋白質磷酸化水平等[12-14]。
雖然,學術界尚未詮釋“睡眠負荷”的全貌,但是對“睡眠負荷”應有的特性達成了一定共識:①“睡眠負荷”在覺醒狀態和睡眠狀態下,發生動態變化的方向是相反的;②“睡眠負荷”需要有狀態依賴性屬性,即變化幅度隨睡眠或覺醒維持時間的推移而增加;③“睡眠負荷”觸發睡眠的生物學效應是“全或無”過程,即覺醒期間積攢的“睡眠負荷”并不是時時刻刻觸發睡眠,而是需在特定的時間點,以非常強有力的作用方式讓機體迅速進入睡眠狀態。
腺苷作為ATP的最終代謝產物,是第一個被廣泛認可的“睡眠負荷”。在20世紀90年代,McCarley團隊發現,實驗動物基底前腦(basal forebrain,BF)和大腦皮質中腺苷含量隨覺醒時間的延長而增加,隨睡眠時間的延長而減少,并證實腺苷通過腺苷A1受體抑制“促覺醒神經元”,通過A2受體興奮“促睡眠神經元”[15-17]。在此基礎上,中國科學院神經科學研究所徐敏研究團隊利用新型遺傳編碼的腺苷探針,進一步發現BF腦區腺苷的釋放主要依賴于谷氨酸能神經元的活動,若選擇性損傷這些谷氨酸能神經元,睡眠的內穩態功能會嚴重受損[18]。上述研究解釋了“睡眠負荷”腺苷在“特定時間”啟動睡眠的可能性,提示谷氨酸能神經元接受某種信息(如生物鐘信息),在“指定時間”活動度增加,引起大量腺苷的同步性釋放,繼而觸發睡眠。但上述推論還有待于進一步驗證。
1.2 睡眠-覺醒蹺蹺板開關模型
2005年,美國哈佛大學醫學院Saper研究團隊提出“睡眠-覺醒蹺蹺板開關理論模型”(簡稱“蹺蹺板模型”,flip-flop model)[19]。該理論模型的核心觀點為睡眠和覺醒之間的更替是由體內睡眠發生系統和覺醒發生系統交替興奮而引起的,這種交替興奮的解剖學結構基礎是這2個系統之間相互存在抑制性纖維投射。因此,當一個系統的活性被觸發時,就會抑制另一個系統的活性并解除另一個系統對自身的抑制,繼而進入正反饋循環,啟動并維持某一種狀態(睡眠或覺醒)。該模型下,睡眠和覺醒的轉換需要依賴于特定的“開關”,該“開關”需要在“指定時間”觸發睡眠發生系統或覺醒發生系統,以此實現狀態轉換。
在“蹺蹺板模型”提出之初,學術界對“睡眠發生系統”和“覺醒發生系統”以及“開關”的概念十分模糊,也較為局限。當時,Saper研究團隊認為,在嚙齒類哺乳動物中,睡眠發生系統可能是下丘腦腹外側視前核(ventrolateral preoptic nucleus,VLPO)的甘丙肽和GABA神經元;覺醒發生系統可能是腦干網狀上行激活系統,包括藍斑核的去甲腎上腺素能神經元、中縫核的5-羥色胺能神經元、腹側被蓋區的多巴胺能神經元、腦橋被蓋核和背側被蓋核的乙酰膽堿能神經元,此外還有下丘腦背側被蓋核的組胺能(histamine,H)神經元,BF的乙酰膽堿能神經元等;而“覺醒開關”是下丘腦穹隆周核的促食欲素能(orexin,OX)神經元。
隨后睡眠醫學研究工作者圍繞“蹺蹺板模型”開展了深入細致的研究,不僅找到了支持該理論模型的實驗室依據,還借助化學遺傳、光遺傳、鈣成像實驗技術,發現了全新的參與睡眠-覺醒調控的神經元和物質。美國加州伯克利大學Dan研究發現,腹側巨細胞網狀核中GABA能神經元和杏仁核中神經緊張素陽性的GABA能神經元通過抑制腦干的單胺系統,促進非快眼動(non-rapid eye movement,NREM)睡眠的發生[20-21]。美國杜克大學Wang課題組發現,下丘腦視上核團中分泌抗利尿激素的一組神經元可被麻醉劑激活,并促進NREM睡眠,該項研究支持了麻醉劑的藥理作用依賴于內源性睡眠調節通路的假說[22]。陸軍軍醫大學胡志安課題組于2018年在Science發表文章,提出丘腦室旁核的谷氨酸能神經元在覺醒控制中發揮重要作用[23]。復旦大學黃志力課題組發現了尾殼核、腹側蒼白球、伏隔核、吻內側被蓋核和嗅結節等參與睡眠與覺醒調控的新核團[24-25]。
此外,近幾年的睡眠醫學研究整合了睡眠“蹺蹺板模型”和“雙過程理論模型”。筆者認為,“蹺蹺板模型”的“開關”對應“雙過程理論模型”的“穩態調節”。“睡眠開關”是“睡眠負荷”,由此想到,體內可能還有對應“覺醒開關”的“覺醒負荷”,撬動機體從睡眠中快速蘇醒,如饑餓感、焦慮感和求知欲等。此外,“蹺蹺板模型”中“開關”發揮作用的“指定時間”對應“雙過程理論模型”的晝夜節律調節,因此“開關”必須接受來自體內生物鐘的信息,使機體的功能同步于外界環境的變化(圖2)。
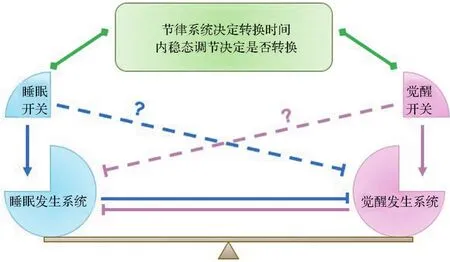
圖2 睡眠-覺醒調控理論示意圖.:調控作用;:興奮作用;:抑制作用.
至今科學家已經發現20余個促覺醒相關腦區和近10個促睡眠相關腦區。但是對絕大多數這些腦區的“促覺醒神經元”和“促睡眠神經元”進行干預(損毀、藥理學方法、光遺傳學激活或抑制等)只影響覺醒或睡眠狀態的水平(時間和程度),并不會誘發覺醒或睡眠狀態的絕對缺失,提示這些睡眠-覺醒相關神經元發揮作用的方式極有可能是一組聯動的、互補的協同效應。以覺醒為例,胡志安教授認為覺醒是覺醒發生系統及其支配的功能網絡共同活動的結果。促覺醒神經元可進一步細分為覺醒發生的“核心組構”和“輔助組構”,“核心組構”決定覺醒的基礎水平,為大腦核心功能網絡提供持續的興奮性輸入,保證大腦不會宕機;而“輔助組構”主要協助大腦核心功能的合理有效運行[26]。睡眠是有別于覺醒狀態的另一種大腦工作模式,因此睡眠發生系統也極有可能通過影響大腦核心功能網絡,對大腦核心功能起不可忽視的關鍵作用,這主要體現在睡眠對記憶的分選作用和對情緒的安撫作用[27-28]。
近年來,隨著神經科學相關研究技術的發展和進步,已經確認了多個可調控睡眠的重要核團,也發現了一些重要的睡眠-覺醒調控的分子生物學機制,但是,至今對“為什么睡覺?”這一問題仍知之甚少。
2 睡眠調節藥物
臨床常見的睡眠疾病包括失眠、睡眠呼吸暫停、日漸過度思睡和猝倒等。睡眠疾病的治療藥物分為促眠藥和促醒藥,主要作用于GABA系統、褪黑素系統、促食欲素系統、組胺系統和單胺系統發揮治療作用。
2.1 作用于GABA系統的睡眠調節藥物
自1958年GABA被認定為抑制性神經遞質以來,科研人員已經發現在中樞神經系統中超過1/3的神經元通過GABA進行信號傳遞。GABA系統紊亂與多種神經系統疾病相關。GABAA受體是配體門控氯離子通道復合物,由α,β和δ等多個不同亞單位組裝成五聚體,其中各亞單位又細分為不同亞型,例如α亞單位有α1~α6 6個亞型。這提示GABAA受體在不同腦區、不同神經元、同一神經元的不同部位以及不同刺激因素下可能呈現不同組裝模式。
除了具有GABA結合位點外,GABAA受體還具有非常重要的苯二氮(benzodiazepine,BDZ)類催眠藥的結合位點GABAA-ω1和GABAA-ω2。GABAA-ω1受體被激活可發揮催眠和抗焦慮作用,而GABAA-ω2受體被激活則出現肌松和認知記憶受損等效應。作為GABAA受體的正向調節劑,BDZ類藥物作用于GABAA受體的BDZ結合位點,使GABA與GABAA受體的親和力增強,更有效地觸發氯離子通道開放,引起細胞膜超極化,產生中樞抑制作用,進而增強睡眠發生系統(如VLPO核團)對覺醒發生系統(如腦干網狀系統)的GABA能抑制性輸入,同時亦在皮質和丘腦等腦區增強GABA的作用,從而發揮催眠作用。然而,這些藥物并不會精準打擊睡眠-覺醒調節通路,而是廣泛地作用于含有GABAA-ω1和GABAA-ω2結合位點的腦區,誘發許多不良反應。BDZ類催眠藥對短期失眠效果較好,可縮短入睡潛伏期,減少覺醒次數,延長睡眠總時間,但縮短快眼動(rapid eye movement,REM)睡眠和慢波睡眠(slow wave sleep,SWS)睡眠,但長期使用可產生依賴性和成癮性。
20世紀60年代以后,BDZ類催眠藥作為常規首選藥物應用于臨床失眠癥的治療,但由于其對GABAA-ω1和GABAA-ω2受體亞基無選擇性,導致不良反應時有發生,促使后續的藥物研發更加注重于具有高選擇性且不良反應更少的鎮靜催眠藥。由此,便出現了新型非苯二氮類催眠藥(Z-drug)。Z-drug選擇性作用于GABAA-ω1結合位點,而對GABAA-ω2無影響(即無認知和記憶功能方面的不良反應)。臨床上常用的Z-drug主要有唑吡坦、扎來普隆和佐匹克隆等,具有起效快、對循環和呼吸系統不良反應少等特點,同時不會引起認知和精神運動障礙,反跳現象輕微。但Z-drug依然具有藥物成癮風險,尤其在精神疾患和藥物濫用人群中,其成癮風險顯著增加[29]。
自2018年開始,Nature相繼報道研究結果解析了不同亞型GABA受體的三維空間結構[30-31]。研究人員采用低溫電鏡技術捕捉到地西泮結合至GABAA受體(α2β2δ和α1β3γ2亞型)后呈現的“活化態”面貌,并闡釋了氟馬西尼競爭性拮抗地西泮的分子動力學過程[32-33]。另有研究表明,呈現βαδββ排列的突觸外GABAA受體中含有一個組胺結合位點,提示體內的組胺和GABA 2種遞質可能共同調節GABAA受體[34]。正如作者提到“GABAA受體的差異化組裝是調節GABAA受體的生理學和藥理學的普遍機制”,當我們獲悉睡眠-覺醒調節通路上的GABAA受體組裝模式,并讓藥物精準打擊具有該模式的受體時,可能會解決GABAA受體正向調節劑面臨的多個臨床痛點。上述研究有望為以GABAA受體為靶點的藥物設計提供重要的理論依據,其中解碼GABAA受體的不同組裝模式與GABAA-ω1和GABAA-ω2藥物結合位點之間對應關系,將會成為非常關鍵的問題。
GABAB受體激動劑用于治療發作性睡病的夜間睡眠紊亂和猝倒。作為γ-羥丁酸的鈉鹽,羥丁酸鈉作用于GABAB受體,通過抑制去甲腎上腺素能神經元、多巴胺能神經元和丘腦皮質神經元,產生中樞抑制作用。羥丁酸鈉可顯著延長深睡眠時間,提高睡眠質量,該效應確保機體在次日白天提高清醒度。羥丁酸鈉在發作性睡病中的成功應用為基于睡眠內穩態調控理論治療覺醒不足的臨床實踐奠定了可行性基礎。在2021年,羥丁酸鈣鎂鉀鈉(JZP-258)和羥丁酸鈉控釋片(FT218)相繼獲得了美國食品藥品監督管理局(FDA)的新藥批復和臨床試驗申請。這些新藥可改善羥丁酸鈉的藥動學特性,降低高鈉引起的心血管風險。這些藥物的成功上市提示,以GABAB受體為靶點的、增加深睡眠的藥物可能對發作性睡病具有獨特的治療作用。近年來,對GABAB受體的結構解析也將有助于基于該靶點的新藥研發[35-36]。
2.2 作用于褪黑素系統的睡眠調節藥物
目前獲批上市的MT受體激動劑有雷美替胺(ramelteon)、特斯美爾通(tasimelteon)和阿戈美拉汀(agomelatine),均屬于促眠藥。MT受體激動劑作用于SCN腦區神經元上的MT1和MT2受體(褪黑素受體),阻斷光信號誘發的促覺醒作用,同時調整機體的生物鐘使其同步于外界晝夜節律,在時差反應、倒班等特殊環境下,促使機體的睡眠-覺醒節律快速適應環境變化。在下午或晚上服用MT或MT受體激動劑可提前睡眠-覺醒時相,晨服則可推遲睡眠-覺醒時相。一項綜合13個臨床試驗、涉及近6000例患者的薈萃分析評估了雷美替胺的效應和安全性,結果表明該藥可顯著縮短主觀睡眠潛伏期,增加睡眠質量,但對主觀總睡眠時間無顯著作用,唯一顯著的不良反應為思睡[37]。特斯美爾通是針對晝夜節律失調性睡眠覺醒障礙研制的藥物。在特斯美爾通的Ⅱ期和Ⅲ期臨床試驗中,受試人員因接受提前5 h的睡眠-覺醒時間安排導致入睡困難,而睡前服用特斯美爾通可縮短睡眠潛伏期、改善睡眠效率和睡眠維持,且無明顯的副作用[38]。阿戈美拉汀相較于前2種的獨特之處在于其同時拮抗5-HT2受體,兼有抗抑郁的作用,因而臨床上可用于治療伴有晝夜節律紊亂的抑郁癥。總體來看,MT受體激動劑對不同原因引起的睡眠疾病均有一定的調節作用,尤其是對睡眠節律障礙的改善,包括睡眠位相滯后障礙和時差反常、倒班作業引起的睡眠疾病。盡管這些藥物的促眠作用比BDZ藥物弱,但由于其不影響學習、記憶和精細運動功能,并且成癮性低,可能更適用于失眠癥的長期治療。
最近研究提示,MT1和MT2受體在睡眠調節中可能具有不同的作用。MT1基因敲除小鼠的NREM睡眠增加,REM睡眠減少,而MT2敲除小鼠則表現為NREM睡眠減少。MT1和MT2受體在睡眠-覺醒相關腦區中的分布不同,MT1受體主要位于藍斑核、下丘腦外側等REM睡眠相關腦區,MT2受體則位于網狀丘腦等NREM睡眠相關腦區。因此,MT1受體可能主要參與REM睡眠調節,而MT2受體主要參與NREM睡眠的調節[39]。由于目前使用的褪黑素受體激動劑對MT1和MT2并無選擇性,因而這類藥物可助于維持睡眠結構的穩定性。但從另一個角度來看,MT受體激動劑無法對REM睡眠和NREM睡眠進行差異化調節,這促使我們思考是否可以單獨針對MT1或MT2受體研制藥物,用于臨床不同睡眠結構障礙的治療。
近幾年對MT1和MT2受體的結構解析加速了MT1或MT2選擇性激動劑的研發工作。2019年,Cherezov研究團隊采用X射線自由電子激光技術,揭示了人MT1和MT2受體的三維結構差異。他們發現MT1的“藥物結合口袋”大于MT2,導致大分子化合物對MT1受體顯示出更高的親和力[40-41]。在此基礎上,采用虛擬分子對接技術,成功找到了具有新穎拓撲結構的選擇性MT1受體可逆激動劑UCSF7447和UCSF3384。在黃昏給予小鼠這些MT1選擇性激動劑,可導致小鼠的晝夜節律時鐘提前1.5 h,該作用在MT1敲除小鼠中被消除,但在MT2敲除小鼠中依然保留[42]。上述研究為選擇性MT受體激動劑的研制工作提供了堅實的理論基礎。
2.3 作用于促食欲素系統的睡眠調節藥物
2022年9月,斯坦福大學的埃馬紐埃爾·米格諾(Emmanuel Mignot)和日本筑波大學的柳沢正史(Masashi Yanagisawa)因發現發作性睡病的發病機制獲得了“2023年科學突破獎”。自1998年這2位科學家獨立發現促食欲素系統功能缺失在發作性睡病中的關鍵作用,到近年3個促食欲素受體拮抗劑蘇沃雷生(suvorexant)、萊博雷生(lemborexant)和達利多雷生(daridorexant)的相繼問世,促食欲素在睡眠-覺醒調節中的作用一直備受關注。
OX是由下丘腦外側穹隆周核神經元分泌的神經肽,并作用于2個興奮性G蛋白偶聯受體——OX1型受體(orexin 1 receptor,OX1R)和OX2R。研究表明,OX2R在睡眠調節中起主導作用,而OX1R對睡眠誘導的貢獻甚小,但其可與OX2R協同調節REM睡眠[43]。目前,獲批上市的蘇沃雷生、萊博雷生和達利多雷生均是OX1R/OX2R競爭性雙重拮抗劑。失眠患者的循證醫學研究也證實了在客觀睡眠參數上OX1R/OX2R雙重拮抗劑的有效性,包括睡眠效率、總睡眠時間和入睡后覺醒的相關指標。并且這些藥物沒有顯示出對記憶和認知的副作用,最常見的不良反應是嗜睡、夢境異常、疲勞和口干[44]。
僅從睡眠-覺醒調節考慮,OX2R似乎是更為理想的干預靶點。事實上,選擇性OX2R拮抗劑的研發已進入臨床試驗階段[45-46]。結果顯示,OX1R/OX2R雙重拮抗劑和選擇性OX2R拮抗劑均可以延長健康受試者的睡眠時間,但對失眠患者睡眠結構的影響不同。OX1R/OX2R雙重拮抗劑主要延長失眠患者的REM睡眠時間,而OX2R選擇性拮抗劑對NREM睡眠和REM睡眠時間均有延長作用[47]。陸林院士曾提出“促食欲素受體將會成為多種精神疾病的治療靶點”,并陳述了促食欲素受體拮抗劑在失眠、抑郁癥、焦慮癥和藥物成癮中的應用進展[48]。促食欲素受體拮抗劑之間的細微差異以及這些藥物在不同疾病中的應用,使我們更加謹慎對待患者睡眠結構的表型分析,并對精神疾患共病性睡眠疾病的診治提出了更高的要求。
另一方面,促食欲素替代品和促食欲素受體激動劑的促覺醒作用也受到了廣泛關注。在促食欲素替代治療中,最棘手的問題是促食欲素替代品難以透過血腦屏障進入中樞分布。發作性睡病動物模型研究結果提示,口服或靜脈注射促食欲素均無效,只有鞘內注射或吸入性給藥方案才能緩解癥狀[43]。因此,基于小分子化合物的促食欲素受體激動劑逐漸成為研發重點。盡管第一款OX2R激動劑YNT-185在臨床試驗中夭折,但對該化合物的研究首次證實了OX2R激動劑在Ⅰ型發作性睡病中的治療作用[49]。由武田制藥研發的OX2R選擇性激動劑TAK-925(靜脈輸注劑型)目前正在Ⅰb期臨床試驗中開展對特發性嗜睡患者的安全性和耐受性評價,其口服劑型TAK-994也已進入Ⅱ期臨床研究并被美國FDA授予突破性療法認定,預期用于治療Ⅰ型發作性睡病和日間過度嗜睡[50-51]。這些藥物的研制有望在發作性睡病和特發性過度睡眠治療中提供新的解決方案。
2.4 作用于組胺系統的睡眠調節藥物
第一代組胺H1受體阻斷劑,如苯海拉明和多西拉敏具有明顯的中樞鎮靜催眠作用,這些藥物可以縮短入睡潛伏期,減少睡眠中覺醒次數。近幾年,具有抗組胺作用的抗精神疾病藥物也逐漸應用于失眠障礙的治療,如多塞平、米氮平、曲唑酮和喹硫平等,小劑量服用該類藥物可產生與阻斷H1受體有關的鎮靜催眠作用。
哌托生特(pitolisant,替洛利生/Wakix)為H3受體拮抗/反向激動劑,是一種新型強效促覺醒劑。哌托生特可通過拮抗組胺H3受體的固有活性,減少組胺H3受體對組胺釋放的負性調節,甚至促進組胺釋放,增強腦部組胺能神經元的活性,表現出顯著的促覺醒作用。哌托生特于2019年被美國FDA批準用于治療發作性睡病成人患者的白天過度嗜睡和猝倒。此外,哌托生特還具有抗疲勞作用和改善認知功能的潛能,使得哌托生特在一些特殊活動如輪班、倒班以及部分軍事活動中具有一定的應用潛力。另一種H3受體反向激動劑SUVN-G3031尚處于Ⅱ期臨床試驗階段[52]。非臨床研究表明,SUVNG3031具有促覺醒作用,且可增加實驗動物大腦皮質的乙酰膽堿、組胺、多巴胺和去甲腎上腺素水平,但并不會升高紋狀體及伏隔核中的多巴胺水平,提示該藥物的成癮風險較低[53]。這與以單胺轉運體為靶點的傳統促覺醒藥(如苯丙胺類)有顯著差別,也是這類藥物的潛在優勢。
2.5 作用于單胺系統的睡眠調節藥物
此類促醒藥多數作用于多巴胺和去甲腎上腺素轉運體,通過抑制單胺類神經遞質的再攝取發揮中樞興奮作用。傳統的單胺類促醒藥為苯丙胺類似物,如苯丙胺、甲基苯丙胺和哌甲酯。近年,新獲批的單胺類促醒藥有莫達非尼(modafinil)、阿莫達非尼(armodafinil)和索安菲特(solriamfetol,JZP-110)。這些促醒藥主要用于治療中樞性過度睡眠,如發作性睡病、特發性過度睡眠、Kleine-Levin綜合征以及藥物或毒品引起的過度嗜睡等,并可有效緩解日間過度思睡。
莫達非尼與多巴胺轉運體結合的親和力較低,因此其藥理作用較為溫和且藥物依賴及濫用的風險較低。阿莫達非尼是外消旋莫達非尼的R-對映體,具有更長的半衰期,最近也被美國FDA批準用于治療發作性睡病相關的日間過度思睡,以及治療經鼻持續氣道內正壓通氣治療者的殘余嗜睡和輪班工作的嗜睡[54]。莫達非尼的促醒作用機制尚未完全闡釋,可能與其調節多種神經遞質和神經肽有關,如谷氨酸、GABA、5-羥色胺、H和OX等[55]。此外,莫達非尼也作為促認知劑在治療認知缺陷相關疾病方面顯示出積極的治療效果,其中包括對工作記憶、情景記憶以及其他依賴于前額葉皮質和認知調控過程的改善[56]。索安菲特是一種多巴胺/去甲腎上腺素再攝取抑制劑,其促覺醒作用機制似乎與調節睡眠-覺醒的其他神經遞質受體(如組胺或食欲素受體)無關,同時因其不釋放單胺類物質而與苯丙胺類興奮劑有所區別。索安菲特于2019年被美國FDA批準用于治療伴有發作性睡病或阻塞性睡眠呼吸暫停的成人患者[57]。
此外,尚有幾個基于該機制的藥物已進入臨床試驗階段。瑞波西汀(reboxetine,AXS-12)是高選擇性、強效去甲腎上腺素再攝取抑制劑。瑞波西汀具有抗抑郁、促覺醒、維持肌張力和增強認知等藥理作用,最初獲批的適應證為抑郁癥。研發人員基于瑞波西汀對去甲腎上腺素系統獨有的強效作用,以“老藥新用”思路,將研發重點切換至發作性睡病。2020年,瑞波西汀獲得了美國FDA頒發的發作性睡病突破性療法認定,但隨后美國FDA又批準了哌托生特用于治療發作性睡病患者的猝倒,因而取消了對瑞波西汀的突破性療法認定。在促食欲素缺乏導致的發作性睡病小鼠模型中,瑞波西汀可以顯著降低猝倒發生率[58]。納入21例發作性睡病患者的多中心Ⅱ期臨床試驗結果表明,瑞波西汀與安慰劑相比顯著降低了治療期間(2周)平均每周的猝倒發作次數,顯著改善患者日間過度思睡癥狀,同時改善了認知功能。該試驗中,瑞波西汀的耐受性良好,最常見的不良反應是焦慮、便秘和失眠[59]。
THN102是另一個進入臨床試驗階段的促醒藥。THN102是莫達非尼和氟卡尼(flecainide)的組合制劑。氟卡尼是一種星形膠質細胞連接蛋白抑制劑。在臨床前研究中,氟卡尼增強了莫達非尼對野生型小鼠的促醒和促認知作用,而莫達非尼和氟卡尼聯合用藥減少了OX敲除小鼠直接過渡到REM睡眠的次數和持續時間,提示聯合用藥方案可能會改善發作性睡病的睡眠結構異常[60]。但是,對發作性睡病伴或不伴猝倒的成年人開展的Ⅱ期臨床試驗結果表明,THN102與莫達非尼單獨治療相比,療效并無差異,因此針對發作性睡病的THN102研發暫時被擱置[61-62]。盡管THN102的新藥研發歷程不盡人意,但是“促醒劑+星形膠質細胞連接蛋白抑制劑”的組合模式給科研工作者帶來新的啟發。考慮到星形膠質細胞和小膠質細胞可能在睡眠期間扮演“大腦清道夫”的角色[63-64],“睡眠調節藥+膠質細胞調節劑”的獨特組合模式有望為神經退行性疾病的治療提供重要的參考意義,如帕金森綜合征[65]。
3 結語
盡管目前學術界尚未完全清晰闡釋“覺醒發生系統(促覺醒神經元)”和“睡眠發生系統(促睡眠神經元)”的神經網絡調控機制,但是對該系統提出了基本的研究框架和理論假設,并達成了一定共識,即晝夜節律對睡眠-覺醒調控系統提供“定時服務”,而內穩態平衡對其提供“驅動力”。此外,近年來睡眠醫學基礎研究成果也在一定程度上推動了睡眠疾病治療藥物的研發,如MT系統藥物、OX系統藥物和H3受體反向激動劑,均為基于睡眠研究理論轉化成功的實例。然而,睡眠疾病表型繁多,病因復雜,未來針對不同病因、不同表型的睡眠疾病治療藥物研發依然任重道遠,尚需睡眠醫學工作者的深耕探索。
猜你喜歡
工業設計(2022年8期)2022-09-09 07:43:20
軍民兩用技術與產品(2021年10期)2021-03-16 06:05:30
北京測繪(2020年12期)2020-12-29 01:33:58
裝備制造技術(2019年12期)2019-12-25 03:06:46
制造技術與機床(2019年10期)2019-10-26 02:47:06
中國洗滌用品工業(2019年4期)2019-05-11 09:27:34
鐵道通信信號(2018年5期)2018-06-28 03:06:24
家庭影院技術(2017年9期)2017-09-26 03:41:45
知識經濟·中國直銷(2017年5期)2017-06-15 20:28:19
通信電源技術(2016年6期)2016-04-20 06:21:32
