
當歸精準煮散飲片指紋圖譜建立及4種成分含量測定
2022-03-07 09:24:28俱蓉楊秀娟李響王本歡權起元李碩靳子明
中國中醫藥信息雜志 2022年2期
俱蓉 ,楊秀娟 ,2,3,李響 ,王本歡 ,權起元 ,李碩 ,2,3,靳子明
1.甘肅中醫藥大學,甘肅 蘭州 730000;2.甘肅省高校中(藏)藥化學與質量研究省級重點實驗室,甘肅 蘭州 730000;3.甘肅省中藥質量與標準研究重點實驗室培育基地,甘肅 蘭州 730000;4.甘肅中醫藥大學附屬醫院,甘肅 蘭州 730000
當歸始載于《神農本草經》,列為中品,來源于傘形科植物當歸(Oliv.)Diels.的干燥根,甘肅所產為“岷歸”,為甘肅道地藥材。現代研究表明,當歸主要活性成分有揮發油(洋川芎內酯Ⅰ、藁本內酯等)、多糖(當歸多糖等)、有機酸(綠原酸、阿魏酸等)等,具有調節機體免疫功能、抗菌、抗氧化等功能,對呼吸、血液、神經等系統有一定作用。
中藥煮散是指原藥材或飲片通過一定的加工方式,得到具有一定規格的粗顆粒,將其在水中煎煮而煎得的湯藥。與傳統湯劑比較,煮散的有效成分含量和干膏率均高于飲片,且中藥煮散在節省藥材、節約煎煮時間及增加煎出率等方面有明顯優勢。宋代龐安時《傷寒總病論》有“湯液之制,遭值天下禍亂之久,地脈薄產之時,天災眾多之世,安得不吝惜而為煮散乎”的描述,可見煮散目的是在資源稀缺時替代飲片使用、節省藥材。但目前煮散“粗顆粒”的粒徑確定尚無科學依據,且煎煮得到的湯劑易粘鍋、有焦味,煮散的形態與傳統飲片相比無法進行準確鑒別,且難以確定煮散與傳統飲片的對應量效關系,亟需對煮散制備工藝及質量控制標準進行研究。精準煮散飲片可為煮散規范化生產及質量控制提供研究思路。當歸精準煮散飲片是指將當歸飲片通過一定加工方式制作成規格統一的粗顆粒。本課題組前期研究得到當歸精準煮散飲片的最佳粒度為 0.400~0.475 cm。
中藥指紋圖譜技術現已廣泛用于中藥材質量控制。利用指紋圖譜可從整體上評價當歸藥材質量,具有體現藥材特異性和信息豐富性的作用。對當歸精準煮散飲片進行指紋圖譜研究,是對其內在質量的保證,也是對其關鍵化學信息的保存。本研究采用HPLC對10批當歸精準煮散飲片進行指紋圖譜研究,并測定綠原酸、阿魏酸、洋川芎內酯Ⅰ和藁本內酯的含量,將傳統飲片與精準煮散飲片作對比,得到2種飲片“量”的對應關系,為當歸精準煮散飲片的質量控制及其開發利用提供參考依據。
1 儀器與試藥
CPA224D電子天平、BSA224S-CW十萬之一電子天平(德國賽多利斯科學儀器有限公司);LC-20高效液相色譜儀、DAD檢測器(日本島津);CH-300型粗碎機(江陰市高宏機械制造有限公司);KQ500VDE超聲波清潔器(昆山市超聲儀器有限公司)。
阿魏酸對照品(批號 L03A9D57744,純度≥98%),綠原酸對照品(批號Y22M8K365444,純度≥98%),藁本內酯對照品(批號R06S10F97166,純度≥98%),洋川芎內酯Ⅰ對照品(批號 A6A10F85918,純度≥98%),上海源葉生物科技有限公司;乙腈為色譜純,甲醇、磷酸為分析純,水為娃哈哈純凈水。
當歸飲片(甘肅隴西保和堂藥業有限責任公司,批號 20191002、20191005、20191006、20191101、20191102、20191104、20191106、20191201、20191204、20191205,編號 S1~S10),經甘肅中醫藥大學李碩副教授鑒定為傘形科植物當歸(Oliv.)Diels的干燥根。將當歸飲片制備為煮散顆粒,備用。本品呈方形、菱形或類方形的不規則狀粗顆粒,粒度為0.400~0.475 cm(4~5目),表面淺棕色至棕黃色,有的具有外表皮,香氣濃郁,味甘、辛、微苦。
2 方法與結果
2.1 色譜條件
色譜柱為Robusta C18(250 mm×4.6 mm,5 μm),流動相為乙腈(A)-0.2%磷酸溶液(B),梯度洗脫程序見表1,檢測波長280 nm,進樣量20 μL,流速1.0 mL/min,柱溫30 ℃。
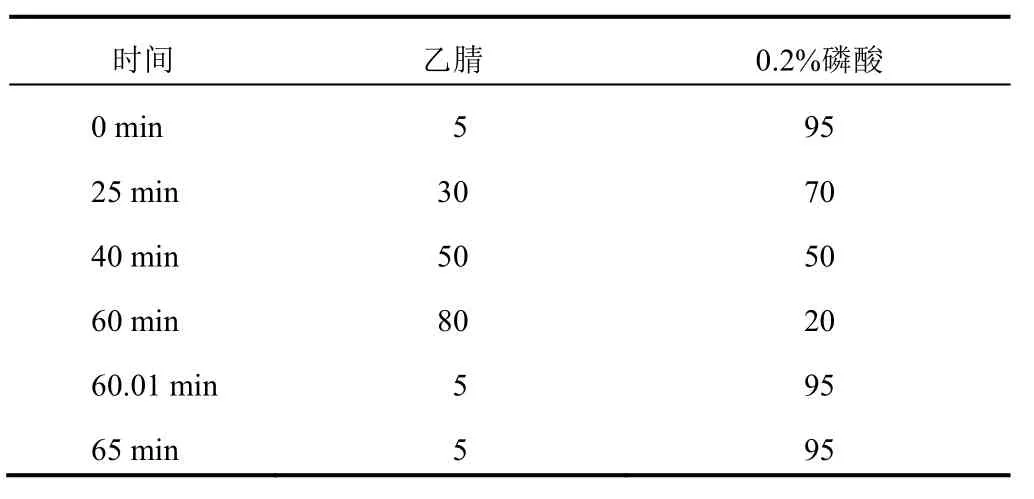
表1 流動相梯度洗脫程序(%)
2.2 對照品溶液制備
精密稱取阿魏酸、綠原酸、洋川芎內酯Ⅰ和藁本內酯對照品適量,加甲醇制成濃度分別為 0.870 4、1.170 1、0.890 0、2.101 1 mg/mL的對照品溶液。分別吸取上述對照品溶液1.00 mL,定容至100 mL棕色容量瓶中,制成混合對照品溶液。
2.3 供試品溶液制備
取當歸精準煮散飲片1 g,精密稱定,置錐形瓶中,加70%甲醇25 mL,稱定質量,超聲(功率250 W,頻率40 kHz)提取40 min,取出,冷卻至室溫,再次稱定質量,用70%甲醇補足減失的質量,過濾,取續濾液,用0.45 μm微孔濾膜過濾,取續濾液,即得。
2.4 指紋圖譜方法學考察
2.4.1 精密度試驗
精密吸取供試品溶液20 μL,按“2.1”項下色譜條件連續進樣6次,采用國家藥典委員會《中藥色譜指紋圖譜相似度評價系統》(2012版)計算,各指紋圖譜與對照指紋圖譜的相似度均大于0.99,表明儀器精密度良好,符合指紋圖譜的要求。
2.4.2 重復性試驗
取同一批樣品,按“2.3”項下方法平行制備 6份供試品溶液,按“2.1”項下色譜條件進樣,采用《中藥色譜指紋圖譜相似度評價系統》(2012版)計算,指紋圖譜與對照指紋圖譜的相似度均大于0.99,表明此方法重復性良好。
2.4.3 穩定性試驗
取樣品,按“2.3”項下方法制備供試品溶液,分別于0、2、4、8、16、24 h,按“2.1”項下色譜條件進樣,采用《中藥色譜指紋圖譜相似度評價系統》(2012版)計算,各指紋圖譜與對照指紋圖譜的相似度均大于0.99,表明供試品溶液在24 h內穩定。
2.5 指紋圖譜建立
2.5.1 特征指紋峰標定
按“2.3”項下方法制備供試品溶液,按“2.1”項下色譜條件分析 10批當歸精準煮散飲片,指紋圖譜見圖1。將檢測數據導入《中藥色譜指紋圖譜相似度評價系統》(2012版),生成當歸精準煮散飲片的對照指紋圖譜及共有模式,手動標識mark峰并進行相關匹配,確定了 26個共有特征峰,其峰面積總和高于總峰面積的90%。將當歸精準煮散飲片HPLC指紋圖譜與傳統飲片色譜圖和混合對照品溶液色譜圖進行比對,確定了4個共有成分,分別為綠原酸(5號峰)、阿魏酸(8號峰)、洋川芎內酯Ⅰ(12號峰)、藁本內酯(25號峰),見圖2。
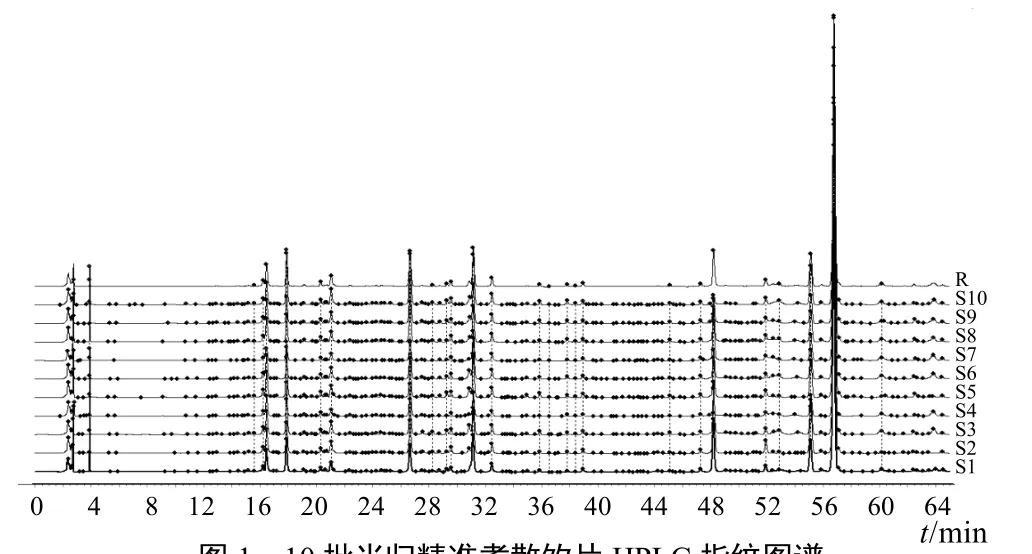
圖1 10批當歸精準煮散飲片HPLC指紋圖譜
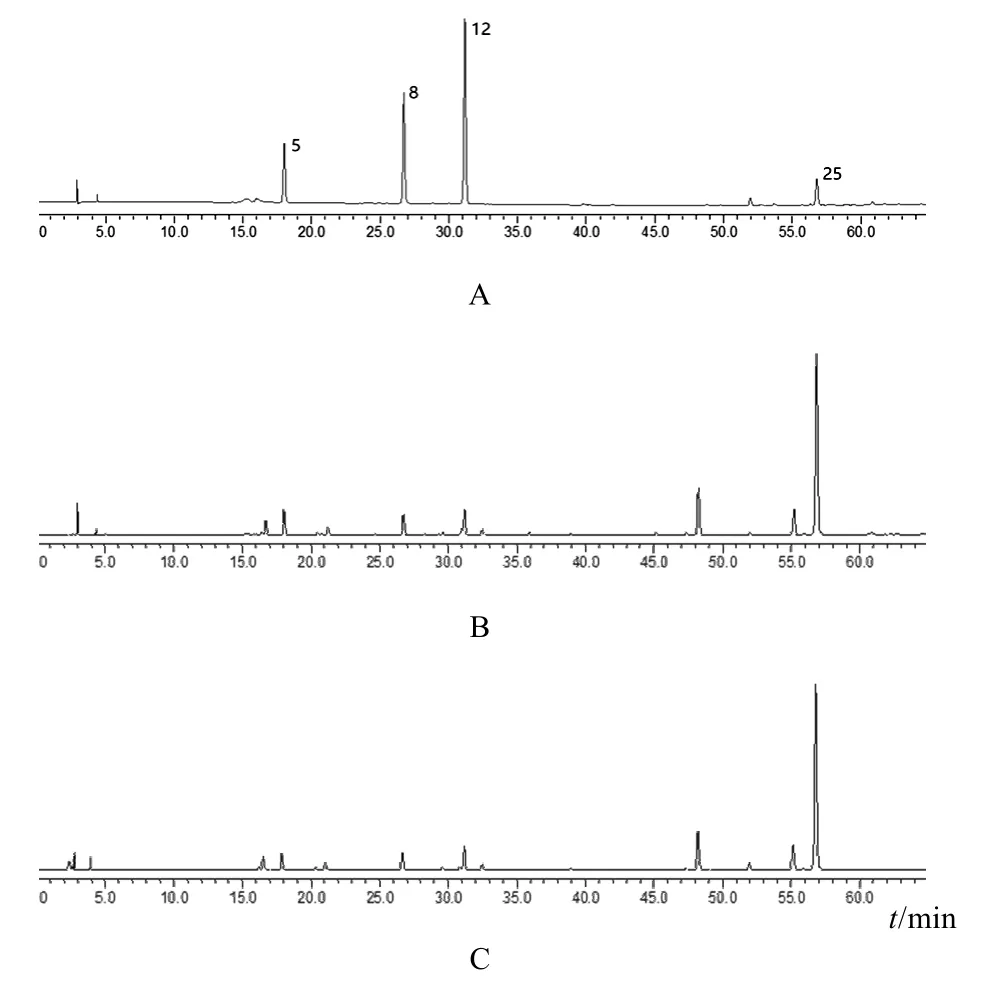
圖2 當歸精準煮散飲片HPLC指紋圖譜特征峰標定
2.5.2 相似度評價
將HPLC指紋圖譜數據導入《中藥色譜指紋圖譜相似度評價系統》(2012版),進行相似度分析,結果相似度均大于0.97,見表2。表明10批當歸精準煮散飲片具有較好的相似性,本試驗建立的指紋圖譜方法體系合理有效。
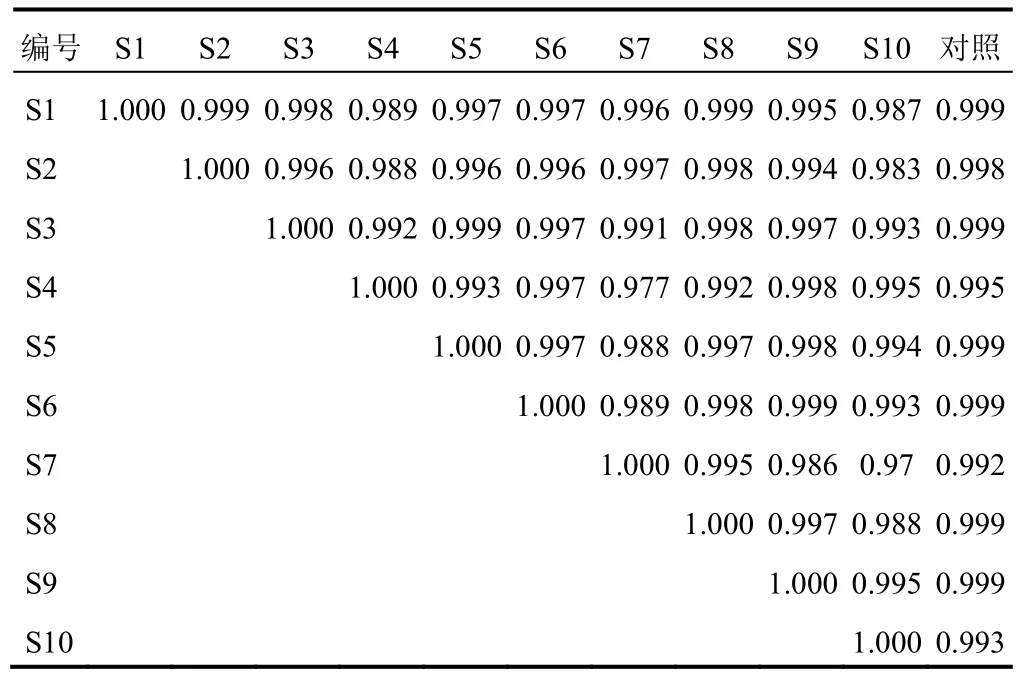
表2 10批當歸精準煮散飲片指紋圖譜相似度評價(r)
2.6 含量測定
2.6.1 線性關系考察
精密吸取“2.2”項下藁本內酯對照品溶液1 mL,用甲醇定容至10 mL棕色容量瓶中。分別精密吸取一定量“2.2”項下綠原酸、阿魏酸、洋川芎內酯Ⅰ和稀釋后的藁本內酯對照品溶液,用0.45 μm微孔濾膜過濾,取續濾液,分別進樣10、20、30、40、60、80 μL。以進樣量(μg)為橫坐標,峰面積為縱坐標,計算綠原酸、阿魏酸、洋川芎內酯Ⅰ和藁本內酯的回歸方程,結果見表3,表明4種成分線性關系良好。
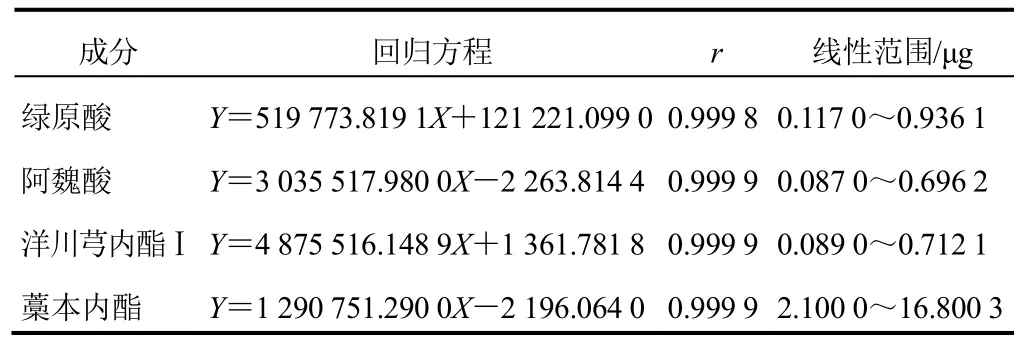
表3 4種成分線性關系考察結果
2.6.2 精密度試驗
精密吸取“2.2”項下對照品溶液,按“2.1”項下色譜條件連續進樣6次,記錄峰面積,得到綠原酸、阿魏酸、洋川芎內酯Ⅰ和藁本內酯的峰面積 RSD分別為 0.92%、0.69%、1.03%、1.79%,表明儀器精密度良好。
2.6.3 重復性試驗
取S1樣品,按“2.3”項下方法平行制備6份供試品溶液,按“2.1”項下色譜條件進樣,記錄峰面積,計算含量,得到綠原酸、阿魏酸、洋川芎內酯Ⅰ和藁本內酯的平均含量分別為 0.473、0.783、0.244、12.331 mg/g,RSD分別為 1.79%、1.18%、1.19%、1.32%,表明該方法重復性良好。
2.6.4 穩定性試驗
取S1樣品,按“2.3”項下方法制備供試品溶液,分別于0、2、4、8、16、24 h,按“2.1”項下色譜條件進樣,結果綠原酸、阿魏酸、洋川芎內酯Ⅰ和藁本內酯的峰面積RSD分別為2.07%、1.18%、1.35%、1.26%,表明供試品溶液在24 h內具有較好的穩定性。
2.6.5 加樣回收率試驗
精密稱取已知含量的樣品6份,分別加入一定量的“2.2”項下對照品溶液,按“2.2”項下方法制備,按“2.1”項下色譜條件測定,計算綠原酸、阿魏酸、洋川芎內酯Ⅰ和藁本內酯的加樣回收率,結果見表4。
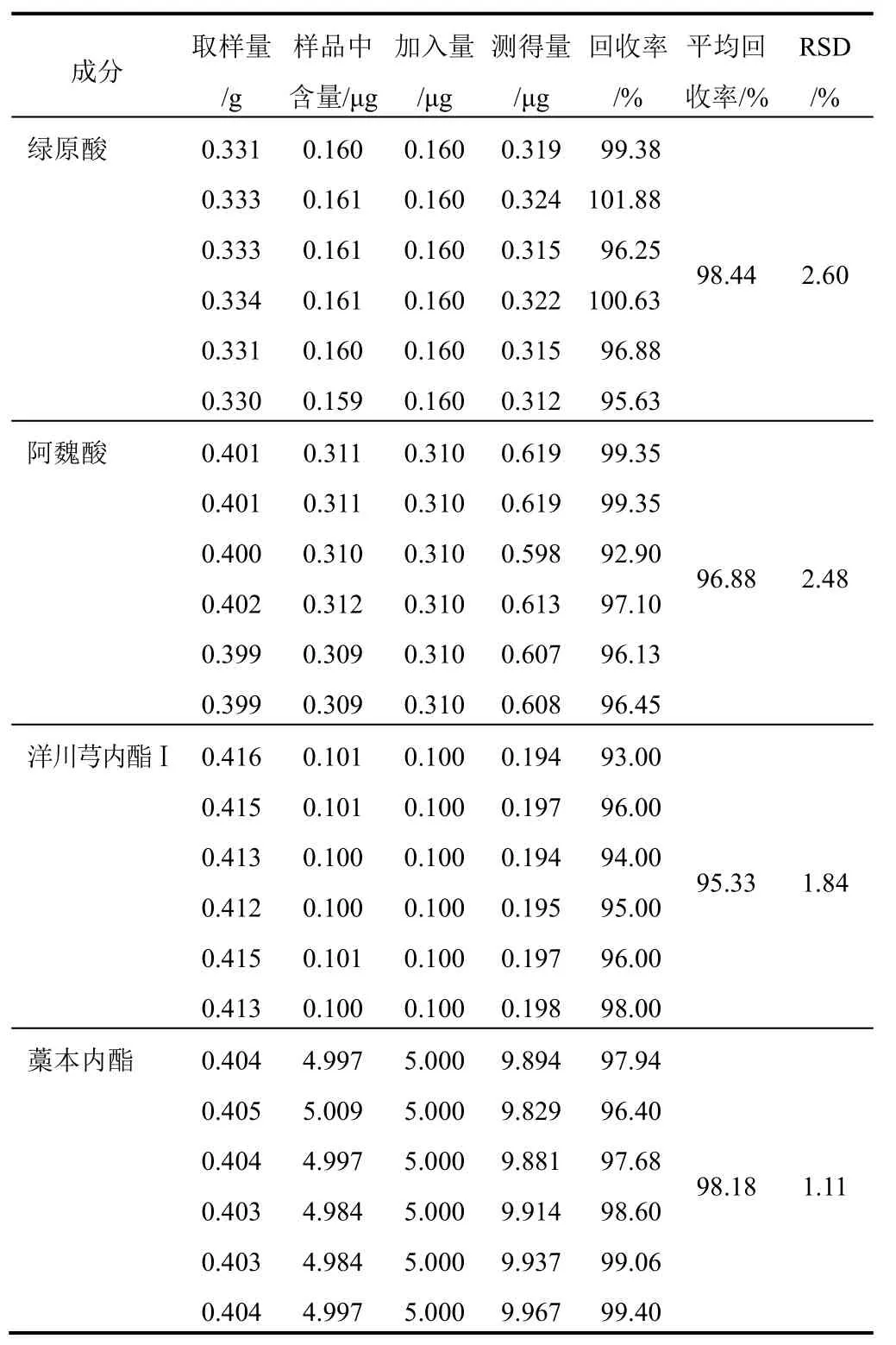
表4 4種成分加樣回收率試驗結果
2.6.6 樣品含量測定
按“2.3”項下方法制備當歸精準煮散飲片和傳統飲片供試品溶液,并按“2.1”項下色譜條件測定,計算 10批當歸精準煮散飲片和傳統飲片中綠原酸、阿魏酸、洋川芎內酯Ⅰ和藁本內酯的含量,結果見表5。
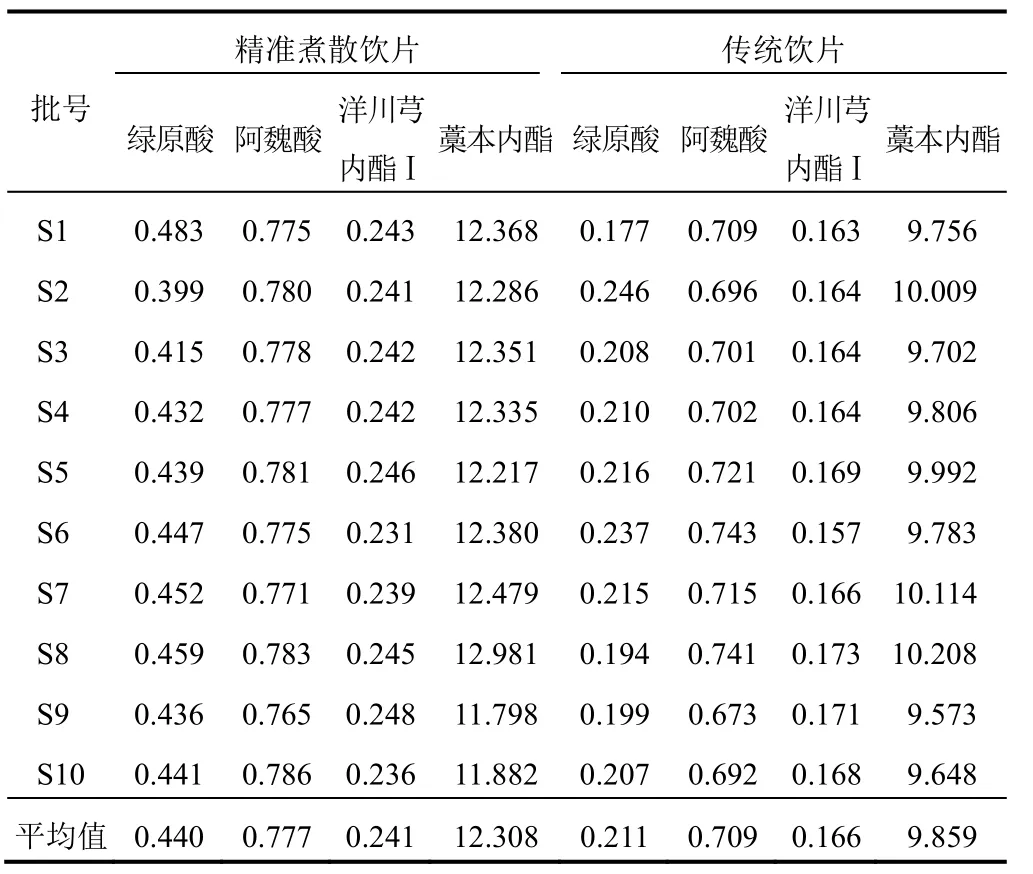
表5 10批當歸精準煮散飲片和傳統飲片中4種成分含量測定結果(mg/g)
2.7 獨立性權重法評分分析
獨立性權重法數據之間相關性,使用回歸分析得到復相關系數R,該值越大表明共線性越強,則權重越低。以 10批當歸精準煮散飲片和傳統飲片樣品中綠原酸、阿魏酸、洋川芎內酯Ⅰ和藁本內酯含量為評價指標,進行加權評分,4種成分的權重見表6。將當歸精準煮散飲片與傳統飲片的4種成分均值與權重系數相乘得到的綜合得分進行比較,得到1 g當歸精準煮散飲片相當于傳統飲片1.26 g,結果見表7。
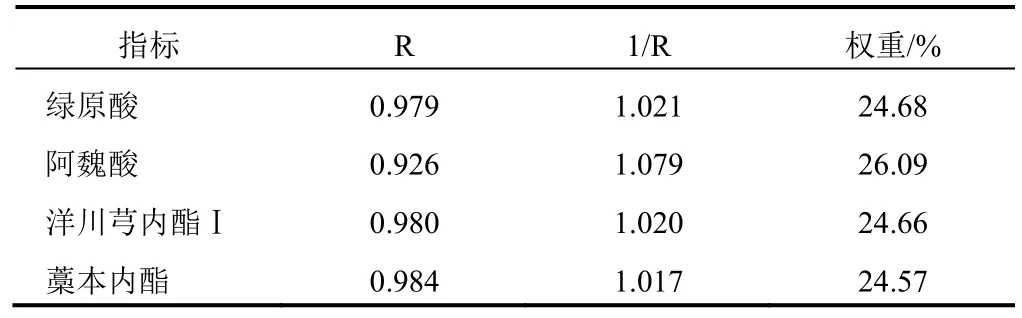
表6 獨立性權重法計算4種成分權重結果
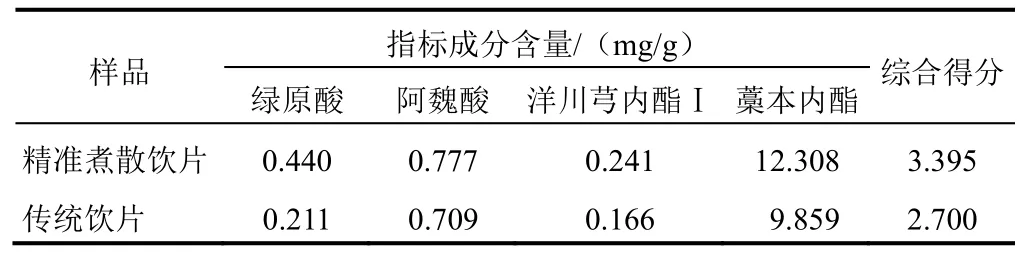
表7 當歸精準煮散飲片與傳統飲片綜合得分比較
3 討論
本課題組在制備當歸精準煮散飲片時,進行了不同粒度的比較,2020年版《中華人民共和國藥典》規定的粉末等級(中粉、細粉、極細粉等)均在煎煮過程中出現了糊化、粘鍋問題,且藥液澄清度不高。考慮當歸精準煮散飲片揮發油隨粒度變小而容易流失,將當歸精準煮散飲片最佳粒度確定為0.400~0.475 cm。精準煮散飲片質量控制的關鍵,是在確定最佳粒度的基礎上評價其整體藥效成分含量,故課題組進行色譜指紋圖譜和代表化學成分含量測定的研究,以期獲得粒度均一、質量穩定的精準煮散飲片。
本研究對色譜條件進行了考察,對乙腈-1%甲酸水、甲醇-水和乙腈-0.2%磷酸作為流動相的洗脫效果進行比較,運用DAD檢測器進行全波長掃描,確定采用乙腈-0.2%磷酸為流動相梯度洗脫,檢測波長為280 nm。將當歸傳統飲片與精準煮散飲片色譜圖進行比對,確定共有成分,10批當歸精準煮散飲片指紋圖譜相似度均大于0.97,表明不同批次間相似度良好,指紋圖譜可代表當歸精準煮散飲片的整體信息。
本試驗在比較CRITIC法、信息量權重法、熵權系數法、獨立性權重法及加權評分法的基礎上,采用獨立性權重法作為綜合評定方法,將每種成分進行單獨加權,結果顯示,當歸精準煮散飲片1 g相當于傳統飲片1.26 g,且當歸傳統飲片中這4種成分的含量整體低于當歸精準煮散飲片。
本試驗建立的指紋圖譜與含量測定方法穩定可靠,可用于當歸精準煮散飲片的質量控制與評價。當歸精準煮散飲片與傳統飲片HPLC指紋圖譜的整體信息,可為其規范化生產及質量控制提供依據,也可為根及根莖類中藥材煮散研究及臨床應用提供參考。
